của phức Host-Guest rất phức tạp. Hiện nay, hằng số bền của loại phức này thường được xác định bằng phương pháp Benesi–Hildebrand. Đây cũng là phương pháp được nhiều nhóm nghiên cứu sử dụng để tính toán hằng số bền của phức giữa ion kim loại với azocalixaren như Chang [34], Le [97], và Ma [107]. Vì vậy, chúng tôi nghĩ rằng phương pháp này phù hợp trong việc xác định hằng số bền của hệ giữa TEAC với Cr(III), Pb(II) và Th(IV) (kí hiệu chung là M).
Tiến hành thí nghiệm như sau: Chuẩn bị 3 dãy bình định mức 25 mL, thêm vào mỗi bình dung dịch ion Cr(III), Pb(II) và Th(IV) sao cho nồng độ ion kim loại là 2.10-4M, rồi thêm dung dịch TEAC 10-3M vào mỗi bình sao cho nồng độ TEAC tăng dần từ 2.10-5 đến 6.10-5M. Điều chỉnh đến giá trị pH tối ưu bằng dung dịch đệm. Lắc đều hệ, sau khi ổn định đo độ hấp thụ quang của hệ TEAC-Pb(II) ở bước sóng 458 nm và hệ TEAC-Cr(III) ở bước sóng 488 nm với dung dịch nền là TEAC cùng nồng độ và hệ TEAC-Th(IV) ở bước sóng 520 nm với nền là dung môi MeOH+H2O. Số liệu được trình bày ở bảng 3.3. Như vậy, bằng phương pháp Benesi–Hildebrand, chúng tôi đã tính toán được hằng số bền của các phức. Các kết quả thu được cho thấy phức của TEAC với các ion Pb(II), Cr(III) và Th(IV) có độ bền cao, có thể ứng dụng để xây dựng quy trình phân tích các ion kim loại này trong mẫu thực.
Bảng 3.3. Hằng số bền (K) và hệ số hấp thụ mol () của hệ TEAC-M
Hệ TEAC-M | K | | |
1 | TEAC-Pb(II) | 4,03.104 | 2,05.104 |
2 | TEAC-Cr(III) | 1,20.104 | 1,42.104 |
3 | TEAC-Th(IV) | 6,14.104 | 2,50.104 |
Có thể bạn quan tâm!
-
 Khảo Sát Tương Tác Của Meac, Deac Và Teac Với Ion Kim Loại
Khảo Sát Tương Tác Của Meac, Deac Và Teac Với Ion Kim Loại -
 Khảo Sát Sự Tương Tác Của Meac, Deac Và Teac Với Ion Kim Loại
Khảo Sát Sự Tương Tác Của Meac, Deac Và Teac Với Ion Kim Loại -
 Khảo Sát Các Yếu Tổ Ảnh Hưởng Đến Sự Hình Thành Hệ Phức Teac- Th(Iv), Teac-Cr(Iii) Và Teac-Pb(Ii)
Khảo Sát Các Yếu Tổ Ảnh Hưởng Đến Sự Hình Thành Hệ Phức Teac- Th(Iv), Teac-Cr(Iii) Và Teac-Pb(Ii) -
 Cấu Trúc Phức Teac-Th(Iv) Tối Ưu Hóa Bằng Arguslab.
Cấu Trúc Phức Teac-Th(Iv) Tối Ưu Hóa Bằng Arguslab. -
 Thứ Tự Các Nguyên Tử Trong Phức Teac- Ion Kim Loại M.
Thứ Tự Các Nguyên Tử Trong Phức Teac- Ion Kim Loại M. -
 Sự Phụ Thuộc Độ Hấp Thụ Quang Của Các Phức Vào Nồng Độ Teac.
Sự Phụ Thuộc Độ Hấp Thụ Quang Của Các Phức Vào Nồng Độ Teac.
Xem toàn bộ 130 trang tài liệu này.
Kết luận phần 3.2
Trong phần này chúng tôi đã khảo sát các điều kiện tối ưu của phức, kết quả thu được như sau:
(1) Phức TEAC-Th(IV) có cực đại hấp thụ tại 520 nm, khoảng pH tối ưu từ 45, thành phần phức 1:1, thời gian ổn định độ hấp thụ quang trong khoảng 590 phút, hằng số bền của phức là 6,14.104 và hệ số hấp thụ mol là 2,50.104 (dung dịch so sánh là MeOH+H2O)
(2) Phức TEAC-Cr(III) có cực đại hấp thụ tại 488 nm, khoảng pH tối ưu từ 1011, thành phần phức 1:1, thời gian ổn định độ hấp thụ quang trong khoảng 590 phút, hằng số bền của phức là 1,20.104 và hệ số hấp thụ mol là 1,42.104 (dung dịch so sánh là TEAC).
(3) Phức TEAC-Pb(II) có cực đại hấp thụ tại 458 nm, khoảng pH tối ưu từ 911, thành phần phức 1:1, thời gian ổn định độ hấp thụ quang trong khoảng 580 phút, hằng số bền của phức là 4,03.104 và hệ số hấp thụ mol là 2,05.104(dung dịch so sánh là TEAC).
3.3. Bàn về cơ chế tạo phức
Đối với các thuốc thử có khả năng tan trong nước và cấu trúc đơn giản thì cơ chế tạo phức của chúng với các ion kim loại thường được đề xuất dựa vào hằng số phân li của thuốc thử, hằng số thủy phân của ion kim loại và các điều kiện tối ưu. Tuy nhiên, phương pháp này gần như không thể áp dụng cho phức Host-Guest, đặc biệt là đối với các loại thuốc thử có cấu trúc phức tạp như azocalixaren. Vì thế, để đưa ra một cơ chế tạo phức phù hợp nhất, chúng tôi đã tiến hành các nghiên cứu sau đây:
- Dùng phương pháp cơ học phân tử (MM) để tối ưu hóa cấu trúc của phức, tính toán độ dài liên kết, góc liên kết, điện tích cân bằng và năng lượng cực tiểu của phức bằng phần mềm ArgusLab 4.01.
- Đo phổ IR của TEAC và TEAC-M để tìm ra sự thay đổi các dao động đặc trưng của các nhóm chức (M là ion kim loại).
- Đo phổ Raman của phức chất để tìm dao động của liên kết MO, MN.
- Đo phổ 1H-NMR để tìm độ dời hóa học của các proton của nhóm OH và nhân thơm khi có mặt của ion kim loại.
- Đo phổ MS để tìm được mảnh ion TEAC-M.
3.3.1. Dự đoán cấu trúc bằng phần mềm ArgusLab
3.3.1.1. Tối ưu hóa cấu trúc phân tử TEAC
Trước hết, dùng công cụ Create New Molecular để xây dựng công thức phân tử của TEAC dạng 2 chiều. Sau đó, sử dụng chức năng Geometry Optimization để tối ưu hóa cấu trúc phân tử. Kết quả thu được biểu diễn ở hình 3.25.
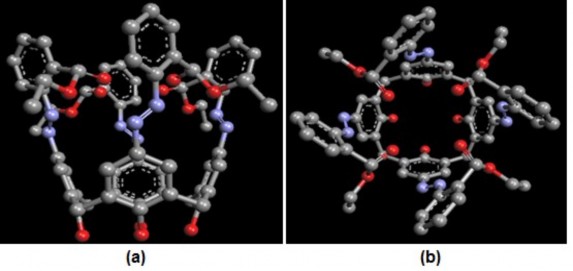
Hình 3.25. Cấu trúc không gian tối ưu của TEAC.
(a) nhìn từ mặt bên; (b) nhìn từ trên xuống
Kết quả tối ưu hóa cấu trúc không gian của TEAC cho thấy bên trong phân tử TEAC có một khoảng không gian trống. Đây chính là nơi các ion kim loại có thể lọt vào để tạo nên phức Host-Guest. Ngoài ra, TEAC còn có các nhóm giàu điện tử như các nhóm azo, nhóm COOEt cũng có xu hướng quay vào bên trong để tạo ra một trường phối tử rất thích hợp cho việc hình thành các liên kết phối trí với ion kim loại.
3.3.1.2. Tính toán mật độ điện tích cân bằng của các nhóm chức
Theo nghiên cứu của Vreven thì mật độ điện tích cân bằng của các nguyên tử trong phối tử là một trong những cơ sở để dự đoán khả năng tạo phức [157]. Vì thế, để dự đoán được khả năng tương tác cũng như vị trí ion kim loại khi xâm nhập vào TEAC, chúng tôi khảo sát điện tích cân bằng của các nguyên tử vùng azo, vùng các nhóm COOEt của vòng trên và các nhóm –OH của vòng dưới với các thông số cài
đặt như sau: Calculation/Geometry/MM/UHF/Calc.QEq Charge/Maximum Steps Taken: 20.000. Kết quả được trình bày ở bảng 3.4 (xem thêm phần phụ lục).
Bảng 3.4. Điện tích cân bằng của một số nguyên tử của TEAC
Dạng azo-enol | Dạng keto-hydrazo | |
N (1) | -0,189 | -0,184 |
N(2) | -0,173 | -0,15 |
N(3) | -0,194 | -0,204 |
N(4) | -0,197 | -0,187 |
N(5) | -0,177 | -0,152 |
N(6) | -0,233 | -0,204 |
N(7) | -0,194 | -0,152 |
N(8) | -0,241 | -0,154 |
O (1) | -0,393 | -0,393 |
O (2) | -0,368 | -0,382 |
O (3) | -0,382 | -0,392 |
O (4) | -0,392 | -0,382 |
O (5) | -0,562 | -0,525 |
O (7) | -0,474 | -0,493 |
O (9) | -0,514 | -0,539 |
O (11) | -0,429 | -0,482 |
Kết quả tính toán điện tích của phân tử TEAC ở cả 2 dạng azo-enol và keto- hydrazo ở bảng 3.4 cho thấy ở khu vực trung tâm của phân tử có 4 nhóm azo với tổng điện tích cân bằng là -1,598 (dạng azo-enol) và -1,387 (dạng keto-hydrazo). Ngoài ra, 4 nguyên tử oxi trong nhóm –C=O của nhóm este cũng có mật độ điện tích là - 1,979 (dạng azo-enol) và -2,039 (dạng keto-hydrazo). Tổng cộng điện tích cân bằng của các nguyên tử N và O có khả năng tạo phức gấp hơn hai lần đối với mật độ electron của các nguyên tử O của vòng dưới (dạng azo-enol là -1,535 và dạng keto- hydrazo là -1,549). Từ các số liệu điện tích cân bằng ở trên, chúng tôi dự đoán vị trí
tạo phức thuận lợi là ở vùng không gian trung tâm. Ngoài ra, TEAC còn 4 nhóm
COOC2H5 ở vị trí octo của các nhân thơm vòng trên nên khả năng các nhóm này có xu hướng xoay vào bên trong khu vực trung tâm hình thành “bẫy” tạo phức Host- Guest với ion kim loại.
Độ dài liên kết và góc liên kết của một số nguyên tử lựa chọn cũng đã được tính toán. Kết quả được trình bày ở bảng 3.5 cho thấy độ dài liên kết giữa các nguyên tử như NN dạng azo nhỏ hơn dạng keto-hydrazo do có sự chuyển dạng liên kết từ đôi sang đơn. Ngược lại, liên kết C(26)-O(4) có độ dài liên kết trong dạng azo-enol lại cao hơn so với dạng keto-hydrazo do đã chuyển từ liên kết đơn sang liên kết đôi. Kết quả này cũng khá tương đồng với số liệu của Ehlinger khi nghiên cứu cấu trúc của azocalixaren tương tự như TEAC [57] (xem cách đánh số thứ tự ở hình 3.36).
Bảng 3.5. Độ dài của một số liên kết trong phân tử TEAC (Ao)
Azo-enol | Keto-hydrazo | Azocalixaren tương tự [57] | |
C(26) - O(4) | 1,407 | 1,279 | 1,398 |
C(23) - N(1) | 1,434 | 1,302 | 1,402 |
N(1) -N(2) | 1,270 | 1,400 | 1,247 |
N(2)-C(57) | 1,434 | 1,434 | 1,402 |
3.3.1.3. Khảo sát hướng xâm nhập và dạng ion kim loại đi vào trong phức
Xét về cấu trúc lập thể, azocalixaren TEAC có dạng hình ống. Do đó, hướng tạo tương tác host-guest thuận lợi là theo trục đối xứng giữa các vòng theo hai hướng từ lower rim hoặc từ hướng upper rim. Trong khi đó, hướng tấn công của ion kim loại vào trung tâm phân tử qua các vòng bên sẽ không thuận lợi do gặp phải sự cản trở không gian của các cầu –CH2 của vòng dưới và nhóm –COOEt của vòng trên (xem hình 3.26).
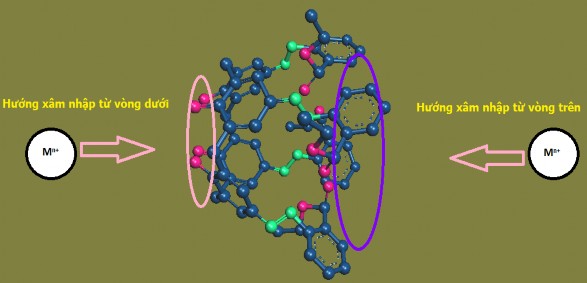
Hình 3.26. Hướng xâm nhập của ion kim loại vào phân tử TEAC.
Đối với phức chất Host-Guest, điều kiện cần để hình thành được phức chất đó là sự phù hợp về kích thước giữa hợp chất “khách” và khoảng trống bên trong phân tử “chủ”. Vì vậy, để phức chất giữa TEAC với các ion kim loại Th(IV), Cr(III) và Pb(II) được hình thành thì kích thước của các ion này ở dạng tự do, dạng hydroxo, hay dạng hydrat của chúng và kích thước các khoang trống ở hai “cửa ngõ” của vòng trên và vòng dưới của TEAC có ý nghĩa quyết định.
Do đó, chúng tôi dùng phương pháp cơ học phân tử để tối ưu hóa kích thước các cation, cách tiến hành như sau: Nhập các công thức của ion kim loại dạng phức hydroxo vào chương trình, tối ưu hóa cấu trúc của các ion này để tìm ra kích thước của chúng. Kết quả thu được cho thấy các ion này có kích thước khá lớn chẳng hạn như ion Pb(OH)3- có kích thước 5,15Ao; Pb(OH)42- 5,21Ao; ion Cr(OH)4- 4,82Ao;
đặc biệt ion Th(OH)3+ có kích thước đến 5,61Ao; Th(OH)22+ 5,54Ao và Th(OH)3+ có
kích thước 3,89Ao. Trong khi đó kích thước các ion Pb2+ 1,02Ao; Cr3+ 0,74Ao và Th4+ là 1,05Ao. Bằng phương pháp tương tự, chúng tôi cũng đã tìm được kích thước vòng trên và vòng dưới của TEAC sau khi tối ưu hóa lần lượt là 4,67Ao và 3,53Ao (hình 3.27).
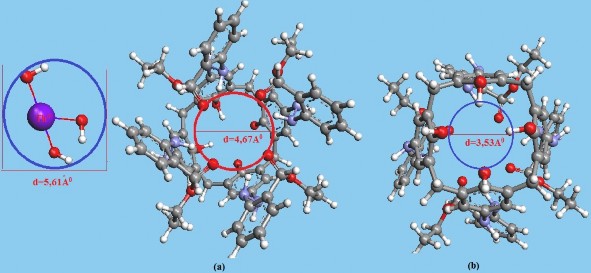
Hình 3.27. Kích thước của vòng trên (a) và vòng dưới (b) của TEAC
Dựa các dữ kiện này, chúng tôi cho rằng các ion kim loại ở dạng phức hydroxo rất khó xâm nhập vào bên trong khoảng trống của TEAC do kích thước của chúng kềnh càng. Vì thế, có thể các ion này tồn tại trong phức chất ở dạng ion kim loại tự do. Hoặc có thể các ion hydroxo này đã bị phân hủy do sự cạnh tranh của các nhóm chức trong TEAC trước ngay khi xâm nhập vào thuốc thử khi đi qua vòng dưới hoặc vòng trên. Năng lượng tối thiểu của các dạng phức hình thành giữa TEAC với các ion hydroxo của ion kim loại cũng đã được khảo sát. Chẳng hạn như trường hợp của phức thori, kết quả nhận được ở bảng 3.6 cho thấy trong 4 dạng
phức được khảo sát, khi thori tồn tại ở dạng ion Th4+ trong phức thì năng lượng của
hệ có giá trị thấp nhất.
Bảng 3.6. Năng lượng cực tiểu của một số dạng phức giữa TEAC và Th(IV)
Dạng phức | Năng lượng cực tiểu (kcal/mol) | |
1 | TEAC-Th4+ | 101,65 |
2 | TEAC-Th(OH)3+ | 121,40 |
3 | TEAC-Th(OH)22+ | 142,90 |
4 | TEAC-Th(OH)3+ | 162,01 |
3.3.1.4. Khảo sát vị trí của ion kim loại trong phức
Kết quả tối ưu hóa cấu trúc phức cho thấy các ion Th(IV), Cr(III) và Pb(II) đều có xu hướng xâm nhập vào lõi trung tâm của phân tử TEAC. Vị trí của chúng gần các nhóm azo và các nhóm cacbonyl của nhóm este. Kết quả khảo sát được trình bày ở các hình 3.28 và 3.29.
Bảng 3.7. Khoảng cách giữa ion kim loại với một số nguyên tử trong phức (Ao)
TEAC-Cr(III) | TEAC-Pb(II) | TEAC-Th(IV) | |
M – N(2) | 2,83 | 2,87 | 2,91 |
M – N(4) | 2,89 | 2,91 | 2,89 |
M – N(6) | 2,98 | 3,04 | 2,98 |
M – N(8) | 2,95 | 3,05 | 2,95 |
M – O(5) | 3,01 | 3,01 | 3,11 |
M – O(7) | 2,94 | 3,03 | 3,03 |
M – O(9) | 2,93 | 2,98 | 3,21 |
M – O(11) | 3,02 | 3,02 | 3,13 |
Khoảng cách giữa ion kim loại với các nhóm tạo liên kết của phối tử cũng được khảo sát. Kết quả ở bảng 3.7 cho thấy khoảng cách giữa ion Cr(III) với nguyên tử N của nhóm azo có giá trị từ 2,82 đến 2,98Ao và Cr(III) với các nguyên tử O của nhóm este là 2,933,02Ao. Khoảng cách giữa ion Pb(II) với nguyên tử N của nhóm azo có giá trị 2,873,05Ao. Trong khi đó, khoảng cách từ ion Th(IV) đến các nhóm chức chứa nguyên tử N và O của TEAC cũng đã được tính toán. Kết quả này cho thấy khoảng cách giữa ion kim loại với các nguyên tố giàu electron như O, N rất phù hợp cho việc hình thành liên kết phức theo quan điểm của Fecman [11].