người. Các nghiên cứu đánh giá dược lý học bao gồm cả cơ chế hoạt động trên động vật sẽ được sử dụng để làm cơ sở đề xuất tiếp tục tiến hành các thử nghiệm lâm sàng.
Lựa chọn động vật thực nghiệm
Việc lựa chọn thử nghiệm trên loài động vật nào để có kết quả tốt nhất là một vấn đề quan trọng hàng đầu trong các nghiên cứu tiền thử nghiệm. Loài thích hợp là một loài mà vật liệu thử nghiệm có tác động dược lý nhờ biểu hiện của cơ quan cảm thụ hoặc yếu tố quyết định kháng nguyên. Một số các kỹ thuật khác nhau có thể sử dụng để xác định loài thích hợp như test chức năng hoặc test hoá miễn dịch.
Các loài động vật để thử nghiệm kháng thể đơn dòng là những loài biểu hiện yếu tố quyết định kháng nguyên mong muốn và cho thông tin về phản ứng chéo trên mô động vật tương tự như trên mô con người. Một loài động vật không biểu hiện yếu tố quyết định kháng nguyên vẫn có thể thích hợp để đánh giá độc tính nếu phản ứng chéo trên mô không định trước với phần tương ứng trên người được chứng minh.
Các nghiên cứu đánh giá tính an toàn thường được thực hiện trên hai loài động vật. Tuy nhiên, trong một số trường hợp ví dụ khi chỉ xác định được một loài thích hợp hoặc khi các đặc tính sinh học của sinh phẩm đã được hiểu đầy đủ thì có thể chỉ cần thử nghiệm trên một loài. Mặt khác có thể cần hai loài để xác định độc tính trong các nghiên cứu ngắn hạn, nhưng chỉ có thể sử dụng một loài để xác định độc tính trong những nghiên cứu dài hạn.
Những nghiên cứu về độc tính trên những loài không thích hợp có thể đưa đến những kết quả sai lệch vì vậy được khuyến cáo không nên sử dụng. Trong trường hợp không có loài thích hợp, người ta có thể sử dụng những động vật biến đổi gen tương thích có các có quan cảm thụ tương tự con người hoặc các protein tương đồng để thực hiện nghiên cứu. Khi không có khả năng sử dụng mô hình động vật biến đổi gen tương thích hoặc các protein tương đồng có thể đánh giá một số đặc tính của độc tính bằng cách lặp lại liều đã dùng trong vòng 14 ngày. Những năm gần đây đã có rất nhiều tiến bộ trong phát triển nghiên cứu trên các mô hình động vật tương tự như con người.
Những mô hình này có thể giúp các nhà nghiên cứu hiểu thấu đáo hơn không chỉ về xác định dược lý học, dược động học và liều lượng mà còn có thể hỗ trợ trong việc xác định tính an toàn của sản phẩm nghiên cứu.
Chọn liều và đường dùng
Có thể bạn quan tâm!
-
 Sơ Đồ Cấu Trúc Hạt Virut Rota Và Các Vùng Mã Hóa Protein
Sơ Đồ Cấu Trúc Hạt Virut Rota Và Các Vùng Mã Hóa Protein -
 Tình Hình Nghiên Cứu Sản Xuất Vắc Xin Rota Trên Thế Giới
Tình Hình Nghiên Cứu Sản Xuất Vắc Xin Rota Trên Thế Giới -
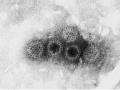
![Hình Ảnh Hạt Virut Rota G1P [8] Dưới Kính Hiển Vi Điện Tử Tại Lần Cấy Truyền Thứ 30 Trên Tế Bào Thận Khỉ Tiên Phát Chụp Dưới Kính Hiển Vi](https://tailieuthamkhao.com/uploads/2022/11/11/danh-gia-tinh-an-toan-va-tinh-sinh-mien-dich-cua-vac-xin-rotavin-m1-do-viet-nam-5-1-120x90.jpg) Hình Ảnh Hạt Virut Rota G1P [8] Dưới Kính Hiển Vi Điện Tử Tại Lần Cấy Truyền Thứ 30 Trên Tế Bào Thận Khỉ Tiên Phát Chụp Dưới Kính Hiển Vi
Hình Ảnh Hạt Virut Rota G1P [8] Dưới Kính Hiển Vi Điện Tử Tại Lần Cấy Truyền Thứ 30 Trên Tế Bào Thận Khỉ Tiên Phát Chụp Dưới Kính Hiển Vi -
 Các Nghiên Cứu Sau Khi Vắc Xin Được Cấp Giấy Phép
Các Nghiên Cứu Sau Khi Vắc Xin Được Cấp Giấy Phép -
 Các Nguyên Vật Liệu, Dụng Cụ Dùng Cho Uống Văc Xin, Lấy Mẫu Và Xét Nghiệm Trong Phòng Thí Nghiệm
Các Nguyên Vật Liệu, Dụng Cụ Dùng Cho Uống Văc Xin, Lấy Mẫu Và Xét Nghiệm Trong Phòng Thí Nghiệm -
 Sơ Đồ Tuyển Chọn Và Phân Nhóm Đối Tượng Giai Đoạn 2
Sơ Đồ Tuyển Chọn Và Phân Nhóm Đối Tượng Giai Đoạn 2
Xem toàn bộ 163 trang tài liệu này.
Đường dùng và số lần dùng càng giống như đường dùng và số lần dùng dự kiến như trong thử nghiệm lâm sàng càng tốt. Dược động học và sinh khả dụng của sản phẩm trên loài được sử dụng và lượng sử dụng an toàn như sử dụng trên người nên được cân nhắc. Trong các trường hợp này, mức độ phơi nhiễm của động vật thí nghiệm tương quan đến mức phơi nhiễm trên lâm sàng nên được xác định cùng với các tác động của khối lượng, độ tập trung, công thức dược phẩm và đường dùng. Tuy nhiên, đường dùng trên động vật có thể khác với đường dùng trên lâm sàng nếu đường dùng bắt buộc phải thay đổi do sinh khả dụng bị hạn chế, những giới hạn do đường dùng hoặc do kích thước/ sinh lý học của loài động vật thí nghiệm. Mức liều dùng khác nhau dựa trên thông tin về mối quan hệ giữa sự đáp ứng của cơ thể và liều dùng. Trong một số trường hợp, sản phẩm thử nghiệm không có hoặc có rất ít độc tính nên không thể xác định liều tối đa. Những trường hợp này cần dựa vào đánh giá một cách khoa học các yếu tố cơ bản để chọn liều.
Tính sinh miễn dịch

Rất nhiều dược phẩm có nguồn gốc công nghệ sinh học giành cho con người lại có đáp ứng miễn dịch trên động vật. Do vậy, cần đo lường kháng thể sản xuất từ cơ thể sau khi dùng sản phẩm nghiên cứu để có thể phiên giải kết quả nghiên cứu. Các đặc tính của kháng thể như hiệu giá kháng thể trên số động vật có đáp ứng, kháng thể có trung hoà hay không trung hoà cũng nên được cụ thể hoá. Ngoài ra các tác động của quá trình tạo kháng thể với các thông số dược động học, tỷ lệ hiện mắc, mức độ nặng, các tác dụng bất lợi... cũng nên được xem xét khi phiên giải số liệu. Trong hầu hết các trường hợp, đáp ứng miễn dịch của động vật với các sinh phẩm rất khác nhau cũng giống như quá trình đáp ứng miễn dịch trên người. Nếu quá trình phiên giải số liệu
không lưu ý vấn đề này thì sẽ không nhận ra ý nghĩa của quá trình đáp ứng miễn dịch. Quá trình sinh kháng thể ở động vật không thể dự báo khả năng hình thành kháng thể ở người vì con người có thể tạo ra các kháng thể dịch thể chống lại các protein giống con người và các kháng thể này tồn tại rất lâu trong cơ thể. Sự xuất hiện đáp ứng quá mẫn mạnh mẽ đối với các protein tái tổ hợp rất hiếm gặp trên người. Do vậy những kết quả nghiên cứu dương tính với các loại dược phẩm có protein trên chuột lang thường không tiên lượng được các phản ứng trên cơ thể người và ít có giá trị khi ứng dụng trên người.
Như vậy dựa vào các thông tin như trên, các công ty dược sẽ quyết định xem dược phẩm mới có đáp ứng yêu cầu khoa học để tiếp tục nghiên cứu phát triển.
1.5.2. Giai đoạn I
Giai đoạn 1 là giai đoạn lần đầu tiên thử nghiệm hoạt chất mới hay công thức mới của thuốc trên người. Thông thường giai đoạn này người ta thường chọn thử nghiệm trên một nhóm nhỏ những người tình nguyện, khoẻ mạnh khoảng 20 – 50 người. Mục đích nghiên cứu giai đoạn 1 nhằm thiết lập đánh giá sơ bộ về tính an toàn, dược động học và dược lực học của hoạt chất trên đối tượng là con người. Giai đoạn này thường được thử nghiệm tại các cơ sở nội trú để bệnh nhân có thể được cán bộ y tế theo dõi liên tục. Các đối tượng nghiên cứu thường được giám sát cho đến khi qua thời gian bán thải của thuốc. Giai đoạn I thường bao gồm cả việc dò liều để xác định liều thích hợp nhất cho con người. Mặc dù giai đoạn I thường được tiến hành trên những người khoẻ mạnh, nhưng vẫn có một số trường hợp vẫn tiến hành trên bệnh nhân mắc bệnh giai đoạn cuối và không có các biện pháp điều trị nào. Những trường hợp ngoại lệ này thường áp dụng ở những bệnh nhân bị ung thư hoặc HIV. Có một số loại thử nghiệm giai đoạn I khác nhau.
1.5.3. Giai đoạn II
Khi tính an toàn đã được xác định ở giai đoạn I giai đoạn II sẽ được tiến hành trên những nhóm đối tượng lớn hơn, tuy nhiên, vẫn trên số lượng bệnh nhân hạn chế khoảng 20 – 300. Mục đích nghiên cứu giai đoạn 2 nhằm đánh giá hoạt động trị liệu, tính an toàn của hoạt chất trên các bệnh nhân, xác định liều sử dụng và chế độ liều thích hợp để đưa ra trị liệu tối ưu cho thử nghiệm lâm sàng. Đôi khi giai đoạn II được chia làm hai giai đoạn IIA và IIB.
- Giai đoạn IIA được thiết kế để đánh giá liều lượng cần thiết.
- Giai đoạn IIB để đánh giá hiệu lực của dược/sinh phẩm.
1.5.4. Giai đoạn III
Giai đoạn III là nghiên cứu thử nghiệm đa trung tâm, ngẫu nhiên, có đối chứng được nghiên cứu trên số lượng bệnh nhân lớn hơn từ 300 – 3.000 hoặc nhiều hơn nữa tuỳ thuộc loại bệnh và sản phẩm nghiên cứu. Mục đích nghiên cứu giai đoạn III nhằm xác định tính ổn định của công thức, tính an toàn/ hiệu quả ngắn hạn và dài hạn của hoạt chất, đánh giá giá trị trị liệu ở mức tổng thể. Nghiên cứu các phản ứng bất lợi thường xuyên xảy ra, phát hiện các đặc điểm đặc biệt của sản phẩm nghiên cứu. Các điều kiện thử nghiệm lâm sàng trong giai đoạn này được tiến hành gần với điều kiện sử dụng thông thường [3,19].
Mặc dù không bắt buộc đối với tất cả các trường hợp nhưng thông thường mỗi loại thuốc, dược phẩm mới cần được thử nghiệm thành công ít nhất hai lần để chứng minh hiệu lực và tính an toàn của thuốc và sau đó được phê duyệt từ các cơ quan có thẩm quyền như Cục quản lý thuốc và thực phẩm (FDA) của Mỹ, Cục quản lý hàng hoá trị liệu (TGA) của Úc và Cơ quan quản lý thuốc Châu Âu (Cộng đồng Châu Âu). Khi thuốc đã thử nghiệm thành công giai đoạn III, kết quả thử nghiệm sẽ được tập hợp thành báo cáo mô tả toàn diện phương pháp, kết quả trên động vật và người, công thức, qui trình
sản xuất và thời hạn sử dụng. Tài liệu này sẽ trở thành bộ tài liệu chính thức cung cấp cho các nước khác xem xét để có thể phê duyệt cho sử dụng trong nước. Hầu hết các thuốc đã trải qua thử nghiệm giai đoạn III có thể đưa vào sử dụng trên thị trường theo tiêu chuẩn của FDA. Tuy nhiên, khi có các tác dụng bất lợi trầm trọng được báo cáo sau khi thuốc/ dược phẩm đã đưa ra thị trường, thuốc hoặc dược phẩm có thể bị rút khỏi thị trường.
1.5.5. Giai đoạn IV
Giai đoạn IV là các nghiên cứu lâm sàng được tiến hành sau khi thuốc đã được lưu hành. Thử nghiệm lâm sàng giai đoạn này được tiến hành trên cơ sở của các đặc tính của sản phẩm đã được phép lưu hành, thông thường dưới hình thức giám sát hậu mãi hay đánh giá giá trị trị liệu hoặc đánh giá các chiến lược điều trị. Thử nghiệm giai đoạn này còn nhằm mục đích giám sát tính an toàn sau khi thuốc được bán trên thị trường. Giám sát tính an toàn được thiết kế để phát hiện những tác dụng bất lợi hiếm gặp hoặc xuất hiện khi sử dụng thuốc lâu dài trên cộng đồng dân cư đông hơn. Phương pháp nghiên cứu trong giai đoạn này có thể khác nhau nhưng sử dụng các tiêu chuẩn khoa học và đạo đức giống với tiêu chuẩn trước khi thuốc lưu hành.
1.6. Các vấn đề cần lưu ý trong thử nghiệm vắc xin trên người [3,24]
Các giai đoạn thử nghiệm vắc xin nhìn chung cũng tuân thủ các giai đoạn quy định thử nghiệm lâm sàng một loại thuốc hoặc dược phẩm mới. Để có một vắc xin được phép sử dụng trên thị trường cũng phải trải qua các giai đoạn thử nghiệm trên động vật và trên người (Hình 1.6). Giai đoạn I và II nhằm đánh giá tính an toàn, và tính sinh miễn dịch trên một nhóm nhỏ đối tượng. Nếu vắc xin đã được chứng minh tính an toàn và sinh miễn dịch giai đoạn I và giai đoạn II, vắc xin sẽ được tiếp tục thử nghiệm trong giai đoạn III để chứng minh hiệu lực. Giai đoạn III phải được thiết kế với những giả thuyết rõ ràng và thực hiện trên
cộng đồng dân cư sẽ sử dụng vắc xin này trong tương lai. Sau khi được cấp giấy phép, giai đoạn IV được tiến hành để giám sát khả năng bảo vệ của vắc xin. Có thể đánh giá khả năng bảo vệ của vắc xin thông qua đánh giá khả năng đáp ứng miễn dịch với vắc xin của cộng đồng sau khi đưa vắc xin sử dụng vào lịch tiêm chủng. Tuy nhiên, một cách chính xác hơn là đánh giá trực tiếp và so sánh giữa đối tượng sử dụng vắc xin và đối tượng không dùng vắc xin để trả lời câu hỏi có phải vắc xin thực sự có hiệu lực bảo vệ và an toàn khi sử dụng trên cộng đồng hay không? Thiết kế sử dụng cho giai đoạn này thường là nghiên cứu mô tả có thể là nghiên cứu thuần tập hoặc ca bệnh chứng.
Mặc dù vậy khi tiến hành các giai đoạn thử nghiệm trên thực tế cũng có
một số các vấn đề cần quan tâm.
Mục đích
Có đáp ứng miễn dịch không?
Có bảo vệ khi nhiễm VR/VK
Có an toàn không?
Có đáp ứng MD không?
Liều thích hợp nhất?
Có an toàn không?
Mức đáp ứng MD?
Hiệu lực có
ổn định?
Có an toàn?
Vắc xin có an toàn trên cộng đồng?
GĐ tiền LS Giai đoạn I Giai đoạn II Giai đoạn III Giai đoạn IV
Đối tượng
Động vật
30-50 người
200-300 người
300-3000 người
Cộng đồng
Hình 1.6. Tóm tắt các giai đoạn thử nghiệm vắc xin
1.6.1. Đề cương nghiên cứu
Để có một cuộc thử nghiệm vắc xin thành công, việc xây dựng đề cương chất lượng và khoa học là yếu tố quyết định. Đề cương giai đoạn I có thể không cần chi tiết bằng đề cương các giai đoạn II, III, và IV nhưng cũng được qui định rõ những thành tố bắt buộc của đề cương. Theo qui định của Bộ luật các Qui định Liên bang - Mỹ, đề cương bắt buộc phải bao gồm các thành tố sau: (1) chi tiết mục tiêu, mục đích của nghiên cứu; (2) tên và địa chỉ của nghiên cứu viên, của các cơ sở nghiên cứu, và của các Hội đồng đạo đức sẽ tham gia đánh giá nghiên cứu; (3) số đối tượng tham gia nghiên cứu và các tiêu chuẩn chọn vào loại ra; (4) thiết kế nghiên cứu và đặc điểm nhóm đối chứng; (6) mô tả kết quả đầu ra được đánh giá như thế nào; (7) mô tả các phương pháp giám sát đối tượng nghiên cứu và phương pháp giảm thiểu các tác dụng bất lợi cho đối tượng. Bên cạnh đó đề cương cũng bao gồm các nội dung về tổng quan và sự cần thiết phải có nghiên cứu, thông tin về bệnh nghiên cứu như đặc điểm lâm sàng, dịch tễ học, những thông tin đã biết về miễn dịch bảo vệ cũng như đánh giá giữa sự cần thiết phải nghiên cứu và những rủi ro của những người tham gia. Các đề cương nghiên cứu cũng cần mô tả rõ loại nghiên cứu như có đối chứng hay không, mù đơn hay mù kép, phương pháp ngẫu nhiên, cỡ mẫu, phân tích kết quả ra sao.
Trong các thiết kế nghiên cứu liên quan đến đối tượng nghiên cứu là trẻ em cần được cân nhắc kỹ lưỡng hơn. Vấn đề đầu tiên cần cân nhắc là tuổi tiêm/uống vắc xin. Điều này hoàn toàn phụ thuộc vào lứa tuổi nào có nguy cơ cao mắc bệnh mà vắc xin có thể bảo vệ. Đối với hầu hết các tác nhân gây bệnh, lý tưởng nhất là cung cấp vắc xin có khả năng bảo vệ khi trẻ càng nhỏ càng tốt. Tuy nhiên, ở giai đoạn đầu đời trẻ được thừa hưởng miễn dịch từ mẹ và có thể làm bất hoạt một số vắc xin từ virut sống như vắc xin sởi. Do vậy vắc xin sởi không thể tiêm ngay trong những tháng đầu. Mặt khác, giai đoạn đầu đời là thời gian trẻ được tiêm nhiều loại vắc xin, nên cần cân nhắc việc thử nghiệm vắc xin mới cùng thời gian với những vắc xin đang sử dụng. Thông thường những thử nghiêm giai đoạn I thường cho trẻ tiêm/uống vắc
xin nghiên cứu cách các vắc xin khác trong vòng 2 tuần để giảm các yếu tố nhiễu về độ an toàn hoặc các đáp ứng miễn dịch với các vắc xin dùng đồng thời. Về tính kinh tế và thực tế, vắc xin mới nên được thiết kế lịch tiêm trùng với các vắc xin thường quy để giảm chi phí và thời gian. Do vậy những đánh giá về tính an toàn và hiệu lực với ảnh hưởng của các loại vắc xin dùng dồng thời sẽ được đánh giá ở giai đoạn II cùng với những cân nhắc về số liều vắc xin trên một trẻ. Thiết kế một đề cương nghiên cứu trên trẻ em cần phải cân nhắc kỹ lưỡng để giảm thiểu những phiền hà cho đối tượng nghiên cứu nhưng vẫn phải đảm bảo thu thập các số liệu cần thiết.
Vấn đề cần quan tâm nữa là đối với các vắc xin sử dụng công nghệ tái tổ hợp ADN hoặc chứa ADN tái tổ hợp vì mặc dù công nghệ ADN phân tử rất chính xác nhưng một số người cho rằng ADN tái tổ hợp sẽ là mối đe doạ môi trường tự nhiên. Do vậy những vắc xin sử dụng công nghệ này trong phần đề cương, cần giải thích những hậu quả đối với môi trường sau khi sử dụng vắc xin này cho người, khả năng sống của các chủng trong vắc xin trong các điều kiện môi trường khác nhau như nước, đất, thực phẩm đặc biệt là có những so sánh với các chủng hoang dại.
Hiện nay có nhiều loại vắc xin sử dụng chủng vi khuẩn hoặc virut sống có thể thải ra môi trường qua đường hô hấp (như vắc xin Rubella) hoặc qua phân (vắc xin tả hoặc vắc xin bại liệt) có khả năng lây nhiễm từ người sang người. Lây truyền virut vắc xin bại liệt đường uống được lưu tâm ngay từ thời gian đầu tiên sử dụng vắc xin vì vắc xin này tạo ra miễn dịch cộng đồng. Ngoài ra lây truyền virut vắc xin hoặc các virut tái tổ hợp trong một số trường hợp có thể gây bệnh do virut vắc xin. Do vậy, đề cương các giai đoạn I và II phải chú trọng việc mô tả biện pháp đo lường khả năng truyền từ người sang người của các loại vắc xin sống.
1.6.2. Lựa chọn đối tượng tình nguyện tham gia nghiên cứu
Các giai đoạn nghiên cứu cần tuyển chọn người những người khoẻ mạnh đặc biệt là giai đoạn I để tránh những yếu tố nhiễu đến đánh giá tính an toàn của vắc xin. Để có thể lựa chọn đối tượng phù hợp với loại vắc xin