Tuy nhiên cách chia của Katoh và Chuong B Do còn chưa được rõ ràng, chưa chỉ rõ đối với từng khoảng nhỏ dữ liệu. Do đó tôi sẽ phát triển tiếp phương pháp của 2 tác giả Chuong B Do và Kazukata Katoh trong khóa luận này.
Trong khóa luận này, ta tập trung nghiên cứu về 4 chương trình sắp hàng đa chuỗi tốt nhất hiện nay là: CLUSTALW, MUSCLE, PROBCONS, MAFFT (bao gồm L-INS-i, E-INS-i, G-INS-i, FFT-NS-1, FFT-NS-2). Ở đây, chúng ta tập trung vào 2 vấn đề tốc độ và điểm chuẩn (benchmark) để đưa ra 2 cây quyết định cho 2 yêu cầu về tốc độ và benchmark.
3.2 Vấn đề tốc độ
Với các phương pháp kể trên, chúng đều có một giới hạn về dữ liệu đầu vào khác nhau. Vấn đề ở đây, là kiểm tra giới hạn của từng phương pháp. Để kiểm tra, chúng ta sử dụng 3 bộ dữ liệu là BAliBASE2[23], BAliBASE3[24] và Pfam-A[25].
Chúng ta chia thành 3 tình huống riêng biệt:
- Dữ liệu với số lượng chuỗi lớn ( > 200 chuỗi).
- Dữ liệu với số lượng chuỗi nhỏ, tổng số amino acid nhỏ.
- Dữ liệu với độ dài của chuỗi quá lớn ( > 2000 amini acid).
3.2.1 Dữ liệu với số lượng chuỗi lớn ( > 200 chuỗi)
Trong trường hợp này, tốc độ đóng một vai trò vô cùng quan trọng. Trong trường hợp này ta có các phương pháp có thể chạy được là: MUSCLE, FFT-NS-1, FFT-NS-2. Những phương pháp này cho kết quả tương đối thấp, nhưng có tốc độ chạy khá cao. Do đó ta sẽ kiểm tra giới hạn của 3 phương pháp này, các test được trích từ bộ dữ liệu Pfam-A ( là bộ dữ liệu chỉ là một tập hợp các chuỗi protein). Với các test có số lượng chuỗi từ 200 đến 500 chuỗi.
Bảng 3: Kiểm tra các MUSCLE, FFT-NS2, FFT-NS1 với các test có số lượng chuỗi từ 200 đến 500 chuỗi.
MUSCLE | FFT-NS-2 | FFT-NS-1 | |
200 – 250 | 28 / 128 / 68.8 | 5 / 12 / 9.4 | 3 / 11 / 8 |
250 - 300 | 63 / 284 / 113.8 | 9 / 17 / 12.6 | 9 / 18 / 11.8 |
Có thể bạn quan tâm!
-

 Các phương sai sắp hàng đa chuỗi nhanh - 1
Các phương sai sắp hàng đa chuỗi nhanh - 1 -
 Các phương sai sắp hàng đa chuỗi nhanh - 2
Các phương sai sắp hàng đa chuỗi nhanh - 2 -
 Cách Giải Quyết Của Chuong B. Do Và Kazutaka Katoh
Cách Giải Quyết Của Chuong B. Do Và Kazutaka Katoh -
 Các phương sai sắp hàng đa chuỗi nhanh - 5
Các phương sai sắp hàng đa chuỗi nhanh - 5 -
 Các phương sai sắp hàng đa chuỗi nhanh - 6
Các phương sai sắp hàng đa chuỗi nhanh - 6
Xem toàn bộ 50 trang tài liệu này.

47 / 183 / 136.8 | 8 / 19 / 13.2 | 7 / 20 / 12.4 | |
350 – 400 | 74 / 339 / 167.6 | 12 / 31 / 18 | 8 / 21 / 12.8 |
400 – 450 | 102 / 604 / 257.4 | 15 / 56 / 26.2 | 13 / 48 / 22.8 |
450 – 500 | 145 / 738 / 372.2 | 18 / 60 / 34 | 16 / 49 / 27.6 |
300 – 350
Kết quả chỉ ra thời gian chạy nhanh nhất, lâu nhất của một test, và thời gian trung bình của các test đó. Từ những số liệu trên ta có thể thấy. MUSCLE chỉ nên chạy với các test dưới 400 chuỗi. Tiếp tục với các phương pháp FFT-NS2 và FFT-NS1, ta có:
Bảng 4: Kiểm tra FFT-NS2 với các dữ liệu có số lượng chuỗi lớn hơn 400
FFT-NS-2 | |
500 – 1000 | 20 / 306 / 90.4167 |
1000 – 2000 | 97 / 454 / 219.455 |
2000 – 3000 | 250 / 535 / 397.727 |
3000 – 4000 | 350 / 631 / 486 |
4000 – 5000 | 497 / 824 / 651.4 |
Dựa vào các số liệu trên, ta có thể thấy FFT-NS-2 chỉ nên giới hạn chạy với các dữ liệu dưới 4000 chuỗi. Trong 2 phương pháp FFT-NS-2 và FFT-NS-1 thì FFT-NS-2 có bước xử lý thô là FFT-NS-1. Do đó FFT-NS-2 có tốc độ chậm hơn nhưng cho kết quả cao hơn FFT-NS-1. Trong các phương pháp được xét, FFT-NS-1 là phương pháp có tốc độ cao nhất, nhưng cho kết quả tồi nhất. Đây là phương pháp chỉ nên sử dụng khi các phương pháp khác đã không thể chạy được.
3.2.2 Dữ liệu với số lượng sequence nhỏ, tổng số amino axit nhỏ
Với trường hợp này, hầu hết các phương pháp đều có thể chạy được, khi đó các điểm đánh giá phải được đặt nên hàng đầu. Chúng ta nên xử dụng các phương pháp có độ chính xác cao như: PROBCONS, L-INS-i, G-INS-i, E-INS-i.
Với PROBCONS, kiểm tra với 2 bộ dữ liệu BAliBASE2[23] và BAliBASE3[24], ta có các thông số như sau:
Bảng 5: Thời gian chạy của PROBCONS theo tống số amino acid
Thời gian chạy | |
0 – 1000 | 2.3 |
1000 – 2000 | 8 |
2000 – 3000 | 18.1 |
3000 – 4000 | 43.3 |
4000 – 5000 | 71.5 |
5000 – 6000 | 103.5 |
6000 – 7000 | 140.6 |
7000 – 8000 | 190.8 |
8000 – 9000 | 250.25 |
9000 – 10000 | 301 |
Từ những số liệu trên có thể thấy, PROBCONS chỉ nên dùng với những bộ dữ liệu nhỏ (có tổng số amino acid nhỏ). Một cách chính xác, khi cần chạy nhanh và chạy chính xác nên giới hạn tổng số amino acid của dữ liệu lần lượt là 7000 và 9000.
Đối với 3 phương pháp L-INS-i, G-INS-i, E-INS-i, nên giới hạn dữ liệu đầu vào ở mức 200 sequences (Theo Katoh [26]).
3.2.3 Dữ liệu với độ dài của chuỗi quá lớn ( > 2000 amino acids)
Đối với các dữ liệu loại này, thì độ phức tạp không gian là một vấn đề quan trọng cần phải xtôi xét đầu tiên trong việc lựa chọn các phương pháp sắp hàng đa chuỗi. Hiện nay, hầu hết các phương pháp sắp hàng đa chuỗi đều hướng đến việc sử dụng thuật toán quy hoạch động với việc sử dụng độ phức tạp không gian là O(L2) (Ở đây, L
là độ dài trung bình của các chuỗi). Đối với các chuỗi đặc biệt dài (> 2000 amino acids) các phương pháp có độ phức tạp không gian tuyến tính O(L) là sự lựa chọn tối ưu để giải quyết vấn đề này. Với các phương pháp đang được xem xét thì CLUSTALW, FFT-NS-2 và FFT-NS-1 là những phương pháp như thế.
3.3 Vấn đề điểm chuẩn (benchmark)
3.3.1 Với các chuỗi có độ tương đồng cao
Độ tương tự (identity), là thuật ngữ chỉ việc mức độ giống nhau của các chuỗi đầu vào. Theo Katoh và Chuong B. Do, với dữ liệu đầu vào có mức độ tương đồng cao ( > 35 % ), thì việc chạy bất cứ chương trình nào cũng không ảnh hưởng quá lớn đến kết quả cuối cùng [26]. Còn việc kiểm tra mức độ tương tự, tôi đã sử dụng một chương trình sắp hàng đa chuỗi có tốc độ cao (cụ thể ở khóa luận này là FFT-NS-1) để tạo ra các chuỗi sắp hàng (có độ dài bằng nhau), sau đó kiểm tra mức độ tương tự với độ phức tạp tuyến tính (O(L) với L là độ dài của chuỗi sau khi sắp hàng).
3.3.2 Với các chuỗi có độ tương đồng thấp
Với các chuỗi có mức độ tương tự thấp (<= 35 % ), các hệ thống tính điểm chuẩn khác nhau đã thống nhất xác định PROBCONS và L-INS-i là các phương pháp cho kết quả cao nhất hiện nay.
Tuy nhiên phương pháp PROBCONS khi chạy với dữ liệu là các chuỗi DNA luôn rất chậm. Do đó, với các dữ liệu là chuỗi DNA ta không nên sử dụng phương pháp PROBCONS.
Nói chung, sắp hàng các chuỗi có độ tương tự thấp được hiểu là 1 trong 3 trường hợp sau:
Trường hợp 1: global homology – tương đồng (homology) trên toàn chiều dài của chuỗi protein
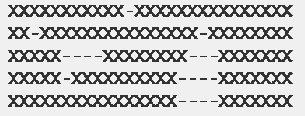
Hình 5: Ví dụ về global homology [4]
Ở đây, X chỉ ra phần có thể được align, o là các phần không được align và – là gap. Theo hình trên ta có thể thấy, toàn chiều dài các chuỗi là các phần có thể được align. Đây là trường hợp đơn giản nhất và phương pháp PROBCONS và G-INS-i là 2 phương pháp cho kết quả tốt nhất trong các phương pháp đang xét.
Trường hợp 2: local homology – tương đồng (homology) được bao quanh bởi các miền không tương đồng
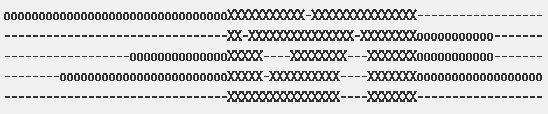
Hình 6: Ví dụ về local homology [4]
Hình trên chỉ ra một tập các chuỗi có chứa trong nó một miền có thể align và xung quanh nó là các phần không tương đồng. Khi đó, L-INS-i là phương pháp tối ưu.
Trường hợp 3: Các đoạn gap nội khối dài - các khoảng tương đồng (homology) ngắn chia tách bởi các đoạn gap nội khối
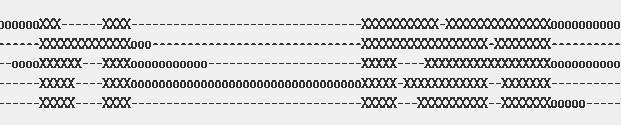
Hình 7: Ví dụ về các đoạn gap nội khối [4]
Trong trường hợp này, có nhiều vùng có thể align, nhưng hầu hết chúng khá rời rạc và được tách bởi những đoạn gap rất dài. Khi đó E-INS-i là phương pháp cho kết quả tốt nhất trong các phương pháp kể trên.
Tuy nhiên, trong hầu hết các hệ thống tính điểm chuẩn, PROBCONS và L-INS-i là hai phương pháp cho kết quả tốt nhất.
3.4 Cây quyết định
Có hai yêu cầu cần phải giải quyết là: tốc độ và benchmark, cho nên ta sẽ tạo hai cây quyết định dựa trên những lý thuyết đã trình bày ở trên.
3.4.1 Cây quyết định cho yêu cầu tốc độ xử lý cao
Hình 8: Cây quyết định với yêu cầu xử lý tốc độ cao
Sequence Input
sequence
>= 2000 aa
Số Sequences
> 4000
Có Không
MAFFT-NS1
MAFFT-NS2
Có
Không
Số Sequences
>= 4000
MAFFT-NS1
Có
Không
Số Sequences
>= 400
MAFFT-NS2
Có
Không
Số Sequences
>= 200
MUSCLE
Có
Không
Tổng số aa
>= 7000
MAFFT-LINSI
Có
Không
Là DNA
MAFFT-LINSI
Có
Không
PROBCONS
3.4.2 Cây quyết định cho yêu cầu tốc điểm chuẩn cao
Hình 9: Cây quyết định với yêu cầu xử lý với điểm chuẩn cao
Sequence Input
sequence
> 2000 aa
Số Sequences
> 4000
Có Không
MAFFT-NS1
MAFFT-NS2
Có
Không
Số Sequences
> 4000
MAFFT-NS1
Có
Không
Số Sequences
> 400
MAFFT-NS2
Có
Không
Số Sequences
> 200
MUSCLE
Có
Không
Độ tương tự
> 35%
MAFFT-LINSI
Có
Không
Tổng số aa
> 9000
MAFFT-LINSI
Có
Không
Là DNA
MAFFT-LINSI
Có
Không
PROBCONS
Trong phương pháp được đề xuất ở đây, tôi chưa xử lý việc tìm cách phát hiện kiểu của các dữ liệu đầu vào có độ tương đồng thấp.
Ở đây ta mặc định với các chuỗi có độ tương tự nhỏ (<= 35 %), chúng ta chỉ sử dụng 2 phương pháp L-INS-i và PROBCONS (là 2 phương án cho kết quả tốt nhất hiện nay). Tuy nhiên kết quả cuối cùng khi chưa xử lý vấn đề này cũng rất khả quan.