- Tìm kiếm và chẩn đoán cấu trúc bậc cao cho chuỗi ADN / protein mới giải mã
- Phân tích phép biến đổi để xây dựng quá trình tiến hóa giữa các loài sinh vật.
- Xác định các vị trí biến đổi dẫn đến các bệnh liên quan đến di truyền, để từ đó tìm ra phương pháp phát hiện và cứu chữa.
Trong quá trình tiến hóa, có 3 phép biến đổi phổ biến trên một trình tự: (1) thay thế, (2) chèn, (3) xóa. Các phép biến đổi này làm cho các chuỗi tương đồng bị biến đổi cả về nội dung cũng như kích thước. Sắp hàng đa chuỗilà quá trình chèn thêm các dấu cách (biểu diễn cho nhưng amino acid bị xóa khỏi chuỗi trong quá trình tiến hóa) vào các chuỗi sao cho tất cả các amino acid ở cùng một ví trí thì tương đồng. Sau khi sắp hàng, tất cả các chuỗi đều có độ dài bằng nhau. Kết quả, ta sẽ thu được một tập các chuỗi gọi là một ‘đa chuỗi thẳng hàng’ ( sequences alignment ).
Ví dụ dưới đây thể hiện một đa chuỗi thẳng hàng của 7 đoạn ADN của 7 loài sinh vật là Người, Mèo, Khỉ, Chó, Ngựa, Gà và Vịt. Phân tích cho thấy ở vị trí thứ 2 tồn tại phép biến đổi giữa ‘C’ của nhóm động vật ( Người, Mèo, Khỉ, Chó ) và ‘G’ của nhóm động vật ( Ngựa, Gà, Vịt ). Tương tự như vậy ta thấy tồn tại các phép biến đổi ở các vị trí 3, 4, 11 và 13. Ở vị trí 7 và số 10, ta quan sát thấy phép biến đổi chèn / xóa ‘G’ và ‘C’ tương ứng.
Bảng 1: Bắt cặp đa chuỗi ADN của Người, Mèo, Khỉ, Chó, Ngựa, Gà và Vịt với các phép thay thế ở vị trí số 2, 3, 11, 13 và phép chén / xóa ở vị trí số 7 và số 10.
1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | |
Người | A | C | A | A | C | T | G | G | T | C | C | G | T | T |
Mèo | A | C | G | A | C | T | G | G | T | C | C | G | T | T |
Khỉ | A | C | G | G | C | T | G | G | T | C | C | G | T | T |
Chó | A | C | G | G | C | T | G | - | T | C | C | G | G | T |
Ngựa | A | G | G | A | C | T | G | G | T | - | C | G | G | T |
Gà | A | G | T | G | C | T | - | G | T | C | G | G | G | T |
Vịt | A | G | T | A | C | T | - | G | T | - | G | G | G | T |
Có thể bạn quan tâm!
-

 Các phương sai sắp hàng đa chuỗi nhanh - 1
Các phương sai sắp hàng đa chuỗi nhanh - 1 -
 Cách Giải Quyết Của Chuong B. Do Và Kazutaka Katoh
Cách Giải Quyết Của Chuong B. Do Và Kazutaka Katoh -
 Dữ Liệu Với Số Lượng Chuỗi Lớn ( > 200 Chuỗi)
Dữ Liệu Với Số Lượng Chuỗi Lớn ( > 200 Chuỗi) -
 Các phương sai sắp hàng đa chuỗi nhanh - 5
Các phương sai sắp hàng đa chuỗi nhanh - 5 -
 Các phương sai sắp hàng đa chuỗi nhanh - 6
Các phương sai sắp hàng đa chuỗi nhanh - 6
Xem toàn bộ 50 trang tài liệu này.
Dễ dàng nhận thấy, chúng ta có thể sử dụng nhiều cách chèn dấu cách vào các vị trí khác nhau để tạo ra các phương án bắt cặp đa chuỗi khác nhau. Trước đây, các nhà sinh vật học có thể tiến hành bắt cặp đa chuỗi bằng mắt và kinh nghiệm. Không cần phải nói cũng có thể hiểu được đó là một công việc vô cùng vất vả và khô khan. Mà kết quả đạt được là rất hạn chế về chất lượng. Qua đó ta có thể thấy được tầm quan
trọng của sắp hàng đa chuỗi. Để nâng cao độ chính xác, các phép biến đổi có thể được gắn các trọng số khác nhau sao cho các phép biến đổi ít khi xảy ra có trọng số lớn hơn các phép biến đổi thường xuyên xảy ra. Đối với dữ liệu protein, người ta thường sử dụng ma trận thay thế axit amin làm trọng số cho các phép thay thế giữa các cặp axit amin ( ma trận thay thế axit amin phản ánh tốc độ thay thế giữa các axit amin ).
1.2 Các chương trình sắp hàng đa chuỗi (multiple sequences alignment ) thông dụng hiện nay
Bài toán sắp hàng đa chuỗi là một trong những bài toán được quan tâm và nghiên cứu nhiều nhất trong hai thập kỉ qua. Một trong các phương pháp nổi bật và thông dụng trước đây là phương pháp CLUSTALW[3] được phát triển bởi Thompson và đồng nghiệp từ những năm 1994. Phương pháp CLUSTALW[3] tiến hành sắp hàng các chuỗi sao cho tổng số điểm phạt (điểm phạt cho phép thay thế, điểm phạt cho phép chèn / xóa…) là nhỏ nhất. Để làm được việc đó, CLUSTALW[3] từng bước tiến hành sắp hàng hai chuỗi (hay hai nhóm chuỗi đã được sắp hàng) để cuối cùng thu được một đa chuỗi thẳng hàng hoàn chỉnh. Tiếp theo CLUSTALW[3], nhiều phương pháp khác đã được đề xuất. Những phương pháp cho kết quả tốt nhất hiện nay là:MAFFT[4], PROBCONS[5], và MUSCLE[6]. Trong đó MAFFT[4] là phương pháp mới được phát triển bao gồm khá nhiều chương trình con cho các yêu cầu khác nhau.
Bảng 2: Các chương trình bắt cặp đa chuỗi phổ biến nhất hiện nay.
Ưu điểm | Nhược điểm | |
CLUSTALW[3] | Tiết kiệm bộ nhớ, có khả năng chạy các test có chuỗi có độ dài lớn. | Kém hơn về độ chính xác và tốc độ so với một số chương trình mới |
MUSCLE[6] | Đạt độ chính xác khá cao và tốc độ nhanh với kích thước dữ liệu vừa phải. | Đối với những tập dữ liệu lớn (>1000 chuỗi), nên chạy với cấu hình tiết kiệm thời gian và bộ nhớ |
PROBCONS[5] | Cho độ chính xác cao khi kiểm tra với một vài bộ dữ liệu chuẩn. | Hạn chế về tốc độ và bộ nhớ, không có khả năng thực hiện với những bộ dữ liệu lớn (>100 |
chuỗi) | ||
MAFFT[4] | Phát triển với nhiều tùy chọn, cho phép thực hiện nhiều yêu cầu từ tốc độ nhanh đến độ chính xác rất cao | Hạn chế về tốc độ với những yêu cầu chạy chính xác, và cũng không phải là phương pháp cho kết quả cao nhất trên tất cả các bộ dữ liệu chuẩn |
Mặc dù việc sắp hàng đa chuỗi và tìm kiếm thành phần lặp có một lịch sử nghiên cứu và phát triển khá lâu, nhưng nó vẫn là một bài toán quan trọng cần phải tiếp tục tập trung nghiên cứu và phát triển để giải quyết các đòi hỏi ngày một cao của lĩnh vực sinh học.
Hàng chục phương pháp sắp hàng đa chuỗi mới được đề xuất hàng năm. Mỗi phương pháp đưa ra đều có ưu điểm và nhược điểm về độ chính xác và thời gian thực hiện. Quan trọng hơn chúng thường chỉ phù hợp cho một số kiểu dữ liệu, và dẫn đến khó khăn lớn cho người dùng trong việc lựa chọn phương pháp phù hợp nhất cho một bộ dữ liệu cụ thể đang nghiên cứu. Ví dụ, đối với các bộ dữ liệu có chứa thành phần lặp, chúng ta phải sử dụng phương pháp tiên tiến nhất cho phép bắt cặp đa chuỗi kết hợp với tìm thành phần lặp. Vì vậy khóa luận sẽ tập trung giải quyết vấn đề trên bằng cách xây dựng một chương trình sắp hàng đa chuỗi kết hợp các phương pháp tốt nhất hiện nay thông qua việc sử dụng cây quyết định.
Chương 2. Các phương pháp bắt cặp đa chuỗi
2.1 CLUSTALW
CLUSTALW[3] là một chương trình được biết đến và sử dụng nhiều nhất trong các chương trình giải quyết bài toán MSA (Multiple sequences alignment). Nó được phát triển bởi Julie D. Thompson, Desmond G. Higgins và Toby J. Gibson.
CLUSTALW[3] được thực hiện thông qua 3 bước chính:
- Tính toán ma trận khoảng cách giữa mọi cặp chuỗi.
- Tạo cây hướng dẫn (guide tree).
- Progressive alignment.
2.1.1 Tính toán ma trận khoảng cách giữa mọi cặp chuỗi
Tại bước này, mọi cặp chuỗi được bắt cặp với nhau, sau đó tính khoảng cách giữa từng cặp chuỗi. Việc này được thực hiện bằng cách sử dụng phương pháp tính toán xấp xỉ nhanh[7]. Phương pháp này cho phép một số lượng lớn các chuỗi được sắp hàng. Còn điểm khoách cách được tính bằng cách: tính số lượng k-tuple khớp với nhau (các đoạn giống hệt nhau, thường có độ dài 1 hoặc 2 cho protein và 2 hoặc 4 cho chuỗi nucleotide) trong kết quả tốt nhất của 2 chuỗi sắp hàng và trừ đi điểm phạt cho việc chèn gap.
2.1.2 Tạo cây hướng dẫn (guide tree)
Từ bước 1, ta có ma trận khoảng cách giữa mọi cặp chuỗi. Cây hướng dẫn (guide tree) cho bước tiếp theo được tạo ra nhờ phương pháp Neighbour-Joining[8]. Đây là một thuật toán lặp. Mỗi lần lặp thuật toán chạy qua các bước sau:
- Căn cứ vào ma trận khoảng cách hiện tại (ở đây là ma trận có ở bước 1) ta tính toán ma trận khoảng cách Q (được định nghĩa dưới đây).
- Tìm các cặp phần tử mà có giá trị khoảng cách Q nhỏ nhất. Tạo nên một nút trên cây và kết hợp 2 phần tử này thành một nút. 2 phần tử này được gọi là “hàng xóm”.
- Tính toán khoảng cách của 2 “hàng xóm” với nút mới.
- Tính toán khoảng cách của các nút bên ngoài với nút mới này.
- Quay lại bước 1 với ma trận khoảng cách được tính từ bước trước.
Thuật toán dừng lại khi chỉ còn lại một nút duy nhất, và nút này trở thành gốc của cây hướng dẫn (guide tree).
Ở đây, ta định nghĩa:
Ma trận khoảng cách ban đầu có r phần tử. d(i, j) là khoảng cách giữa i và j trong ma trận đó.
Khi đó khoảng cách Q giữa i và j được tính:
r r
Q(i, j) (r 2)d (i, j) d (i, k ) d ( j, k )
k 1 k 1
Với mỗi “hàng xóm” khi được nối tạo thành một nút mới, chúng ta sử dụng công thức sau để tính khoảng cách giữa từng “hàng xóm” với nút mới. Ở đây: f, g là 2 hàng xóm và u là nút mới được tạo thành:
1 1 r r
d ( f , u) d ( f , g) [ d ( f , k ) d (g, k )]
2 2(r 2)
k 1 k 1
Khi một nút mới được tạo thành ta dùng công thức sau để tính khoảng cách của nó với các nút cũ:
d (u, k) 1 [d ( f , k ) d ( f , u)] 1 [d (g, k ) d (g, u)] 2 2
Ở đây, u là nút mới, k là nút cũ, f và g là 2 phần tử tạo nên nút mới u.
2.1.3 Progressive alignment
Dựa vào cây hướng dẫn (guide tree) được tạo ra từ bước 2, chúng ta sử dụng sắp hàng các chuỗi từ nút lá cho đến gốc của cây. Mỗi bước sẽ là quá trình sắp hàng 2 nhóm chuỗi đã được sắp hàng trước đó sử dụng thuật toán quy hoạch động [9] [10]. Gap ở những nhóm chuỗi này được giữ nguyên trong kết quả tạo thành. Việc này lặp đi lặp lại cho đến khi gặp gốc của cây hướng dẫn. Đó là kết quả cuối cùng.
2.2. MUSCLE
2.2.1 Các loại khoảng cách và các cách xây dựng cây hướng dẫn
MUSCLE[6] sử dụng hai cách để xác định khoảng cách giữa các chuỗi đó là khoảng cách K-mer[11] cho những cặp chuỗi chưa được sắp hàng và khoảng cách Kimura[12] cho những cặp đã được sắp hàng (có độ dài bằng nhau).
K-mer[11] được định nghĩa là một chuỗi có độ dài bằng K của các amino acid đứng liền kề nhau. Thuật toán sử dụng K-mer không đòi hỏi cặp chuỗi cần phải sắp hàng mà ưu điểm lớn của nó là kết quả thu được với tốc độ khá cao (độ phức tạp thuật toán là O(L) với L là độ dài của chuỗi) [11].

Hình 1: Ví dụ về k-mer [6]
Hình 1 thể hiện một ví dụ của K-mer, với K = 3, ở các chuỗi trên dễ dàng nhận thấy K-mer là 6 và tại các chuỗi ở phần dưới K-mer là 13. Tương tự như vậy với K = 4 các chuỗi bên trên K-mer là 4 và các chuỗi dưới K-mer là 9.
Khoảng cách Kimura[12] giữa 2 chuỗi đã được sắp hàng (có độ dài bằng nhau) được tính theo công thức sau:
DK2p= -1/2 ln(1-2P-Q) – ¼ ln(1/2Q)
Ở đây, P là số lượng transition và Q là số lượng transversion trong hai chuỗi. Transition là một phép thay thế khi thay A bởi G, C bởi T hoặc ngược lại. Trong khi đó Transversion là phép thay thế A bởi C hoặc T hay các trường hợp tương tự.
Sau khi xây dựng xong ma trận khoảng cách, MUSCLE[6] sử dụng thuật toán UPGMA[13] để nhóm các chuỗi lại với nhau. UPGMA[13] là một thuật toán xây dựng cây hướng dẫn cho việc sắp hàng từng profile. Đầu vào của UPGMA[13] là một ma trận khoảng cách. Ở mỗi bước 2 nút gần nhất được nhóm lại với nhau tạo thành một nút mới và ma trận khoảng cách được tính lại theo công thức sau:
1d (x, y)
| A | * | B | xA xB
Ở đây, chúng ta tính khoảng cách giữa 2 nút A và B; x và y lần lượt là các nút ban đầu trong A và B; d(x, y) là khoảng cách giữa 2 nút x và y.
2.2.2 Profile alignment
Để áp dụng bắt cặp sóng đôi vào các profiles, điều cần thiết là xác định một hàm tính điểm cho một cách sắp hàng.
Ta coi i, j là 2 amino acid, pi là xác xuất i xuất hiện, pij là xác xuất i và j được align với nhau, fxi là xác suất của i xuất hiện tại cột x của profile thứ nhất, fxG là xác suất xuất hiện một gap tại cột x trong các profile. Khi đó MUSCLE[6] đưa ra hàm tính Log-Expectation theo công thức sau:
LExy = (1 - fxG)(1- fyG) log ∑i∑j fxi fyi pij/pipj
MUSCLE[6] sử dụng các tham số pi và pij là các tham số của ma trận 240 PAM VTML [14].
2.2.3 Thuật toán
Thuật toán MUSCLE[6] là một loạt các bước sử dụng các khái niệm đã trình bày ở trên. Tuy nhiên tổng quát lại nó bao gồm 3 phần chính là:
Phần 1: draft progessive. Phần 2: improved progessive. Phần 3: Refinement.
Mỗi phần làm một nhiệm vụ riêng biệt, nhưng kết nối chặt chẽ với nhau bởi đầu ra của phần này là đầu vào của phần tiếp theo.
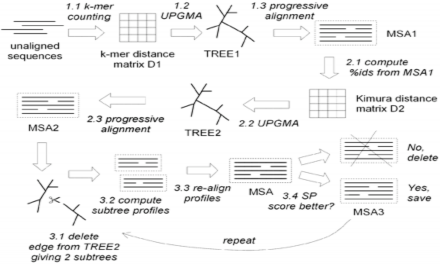
Hình 2: Các bước thực hiện của MUSCLE [6]
Phần 1: draft progessive. Mục tiêu chính của phần này là tạo ra một đa chuỗi thẳng hàng mà vấn đề chính là tốc độ chứ không phải là sự chính xác.
- Sử dụng khoảng cách K-mer[8] để xác định khoảng cách giữa mỗi cặp của chuỗi dầu vào. Tạo ra ma trận khoảng cách D1.
- Sử dụng thuật toán UPGMA[13] để xây dựng cây hướng dẫn TREE1.
- Ở mỗi lá của cây Tree1, một profile được tạo ra từ một chuỗi đầu vào. Các nút trong cây được thăm theo thứ tự tiền tố (nghĩa là các nút con được thăm trước cha mẹ). Ở mỗi nút trong (internal node) phương pháp một bắt cặp sóng đôi (pairwise alignment - một đa chuỗi thẳng hàng nhưng chỉ có được xây dựng từ 2 profile) được xây dựng từ 2 nút con. Việc này lặp đi lặp lại cho đến khi gặp gốc của cây. Đó chính là đa chuỗi thẳng hàng – mục tiêu của bước này.
Phần 2: improved progessive. Hầu hết các lỗi trong phần một đều do việc sử dụng khoảng cách K-mer[11], phần 2 sẽ tạo lại cây bằng cách sử dụng khoảng cách Kimura[12]. Phương pháp này cho kết quả tốt hơn nhưng đòi hỏi các chuỗi phải được sắp hàng (có độ dài bằng nhau).
- Với mỗi cặp chuỗi trong MSA1, chúng ta tính khoảng cách cho chúng sử dụng khoảng cách Kimura[12]. Kết quả ta có ma trận khoảng cách D2.
- Từ ma trận khoảng cách D2, sử dụng phương pháp UPGMA[13] ta tạo nên cây Tree2.