Dựa theo các công bố trước đây xác định được 60 isoflavone có khả năng ức chế HER2 [58-65]. Các hợp chất được tải về từ cơ sở dữ liệu Pubchem (http://pubchem.ncbi.nlm.nih.gov/). Đây là cơ sở dữ liệu lớn nhất thế giới có thể truy cập thông tin miễn phí của các hợp chất hóa học, duy trì bởi Trung tâm Thông tin Công nghệ sinh học Quốc gia, thuộc Thư viện Y khoa Quốc gia Hoa Kỳ.
2.1.3. Cấu trúc chất chứng
Trastuzumab, Lapatinib, Tucatinib là những thuốc chống ung thư, đã được FDA (Cục quản lý Thực phẩm và Dược phẩm Hoa Kỳ) phê duyệt trong điều trị ung thư vú dương tính HER2 [35,44,66] . So sánh docking scores của các hợp chất Isoflavone với các chất này để đánh giá và sàng lọc khả năng ức chế enzym HER2.
2.1.4. Thiết bị và phần mềm
Thiết bị sử dụng: Máy tính để bàn thế hệ core i5-10500 và GTX 1660.
Hệ điều hành Windows 11.
Phần mềm: Trong nghiên cứu sử dụng các phần mềm và công cụ sau:
- MGL tools 1.5.6 (http://mgltools.scripps.edu/)
- Autodock Vina 4.2 (http://vina.scripps.edu/)
- UCSF Chimera 1.15 (https://www.cgl.ucsf.edu/)
Có thể bạn quan tâm!
-

 Đánh giá tác dụng ức chế enzym her2 của các hợp chất isoflavone trong định hướng điều trị ung thư vú bằng phương pháp docking phân tử - 1
Đánh giá tác dụng ức chế enzym her2 của các hợp chất isoflavone trong định hướng điều trị ung thư vú bằng phương pháp docking phân tử - 1 -
 Đánh giá tác dụng ức chế enzym her2 của các hợp chất isoflavone trong định hướng điều trị ung thư vú bằng phương pháp docking phân tử - 2
Đánh giá tác dụng ức chế enzym her2 của các hợp chất isoflavone trong định hướng điều trị ung thư vú bằng phương pháp docking phân tử - 2 -
 Nguyên Liệu, Thiết Bị, Nội Dung Và Phương Pháp Nghiên Cứu
Nguyên Liệu, Thiết Bị, Nội Dung Và Phương Pháp Nghiên Cứu -
 Thể Hiện Tương Tác Trastuzumab Với Protein 3Pp0. Các Chất Chứng Cũng Cho Thấy Khả Năng Liên Kết Với Một Số Acid Amin Quan Trọng Trong Nhóm Lys753, Val734,
Thể Hiện Tương Tác Trastuzumab Với Protein 3Pp0. Các Chất Chứng Cũng Cho Thấy Khả Năng Liên Kết Với Một Số Acid Amin Quan Trọng Trong Nhóm Lys753, Val734, -
 Đánh giá tác dụng ức chế enzym her2 của các hợp chất isoflavone trong định hướng điều trị ung thư vú bằng phương pháp docking phân tử - 6
Đánh giá tác dụng ức chế enzym her2 của các hợp chất isoflavone trong định hướng điều trị ung thư vú bằng phương pháp docking phân tử - 6 -
 Đánh giá tác dụng ức chế enzym her2 của các hợp chất isoflavone trong định hướng điều trị ung thư vú bằng phương pháp docking phân tử - 7
Đánh giá tác dụng ức chế enzym her2 của các hợp chất isoflavone trong định hướng điều trị ung thư vú bằng phương pháp docking phân tử - 7
Xem toàn bộ 57 trang tài liệu này.
- Avogadro 1.2.0 (http://avogadro.cc/)
- Discovery Studio 2021 Client (https://discover.3ds.com/)
- Microsoft Office 2016.
- Microsoft Excel 2016.
2.2. Nội dung nghiên cứu
Bước 1: Sàng lọc các hợp chất isoflavone được tải từ cơ sở dữ liệu Pubchem có khả năng ức chế enzym HER2 bằng phương pháp docking phân tử. So sánh năng lượng với các chất chứng để chọn lọc ra các phức hợp ligand-protein có năng lượng liên kết phù hợp.
Bước 2: Nghiên cứu đặc điểm giống thuốc của các hợp chất có kết quả sàng lọc docking phân tử tốt nhất.
2.3. Phương pháp nghiên cứu
2.3.1. Mô phỏng protein docking
2.3.1.1. Re-dock 03Q
Re-dock là bước quan trọng để thẩm định quy trình docking. Trong protein 3pp0 có chứa sẵn ligand đồng kết tinh là 03Q, Hình 2.2 thể hiện cấu trúc 2D của 03Q. Ligand đồng kết tinh này được re-dock lại vào vị trí hoạt động của protein. Nếu giá trị độ lệch bình phương trung bình gốc (RMSD) nhỏ hơn hoặc bằng 1,5 Å thì có thể kết luận tính phù hợp của quy trình docking [67]. Các bước thực hiện re-dock:
Chuẩn bị protein: Cấu trúc tinh thể của 3pp0 được tải từ Protein Data Bank. Loại bỏ phân tử nước, ligand đồng kết tinh bằng phần mềm Discovery Studio. Tiếp theo sử dụng Autodock Tools 1.5.6 để thêm hydro, tối ưu hóa các hydro phân cực, gắn trường lực Kollman. Vùng hoạt động của protein được xác định trong một hộp lưới có kích thước 30 Å x 30 Å x 30 Å, trung tâm (x,y,z) là (34, 46, -12) [68]. Sau đó lưu protein dưới định dạng pdbqt để chuẩn bị cho quá trình docking.
Chuẩn bị ligand đồng kết tinh: Mở protein 3pp0 bằng phần mềm Discovery Studio. Loại bỏ nước, protein và chỉ giữ lại các phân tử khác. Lưu file dưới đuôi .pdb. Chuyển ligand sang đuôi .pdbqt bằng phần mềm Autodock Tools.
Hình 2.2. Cấu trúc 2D của ligand đồng kết tinh 03Q.
Re-dock: Dữ liệu các file pdbqt đã được xây dựng của ligand đồng kết tinh và protein được tiến hành docking bằng Autodock Vina. Số vòng lặp cho chương trình là 8 vòng. Tính toán giá trị RMSD giữa 03Q khi được tách ra và sau khi thực hiện re-dock bằng phần mềm Chimera.
2.3.1.2. Mô phỏng protein docking với 60 phối tử và chất chứng
Chuẩn bị protein: protein trong phần docking cũng được loại bỏ phân tử nước, ligand đồng kết tinh bằng Discovery Studio; thêm hydro, tối ưu hóa các hydro phân cực và gắn trường lực Kollman và xây dựng file pdbqt bằng Autodock Tools như thực hiện trong re-dock
Chuẩn bị ligand và chất chứng: Cấu trúc 3D của các hợp chất này được lấy từ cơ sở dữ liệu PubChem ở định dạng sdf sau đó chuyển thành định dạng pdb bằng phần mềm Chimera [69,70]. Tiếp theo, các phối tử được tối ưu hóa bằng phần mềm Avogadro sử dụng phương pháp Gradient liên hợp (Conjugate Gradients) rồi chuyển thành định dạng pdbqt bằng phần mềm Autodock Tools 1.5.6 [71,72].
Docking: Dữ liệu các file pdbqt đã được xây dựng của phối tử và protein được tiến hành docking bằng Autodock Vina. Phần mềm được sử dụng để tìm ra cấu hình liên kết tốt nhất bằng cách sử dụng các đánh giá năng lượng tự do liên kết
∆G và số lượng tương tác vật lý. Kết quả docking được đánh giá thông qua 3 tiêu chí là điểm số docking, khả năng tương tác và RMSD (độ lệch bình phương trung bình gốc).
Trong mô phỏng docking, năng lượng liên kết càng thấp được cho là càng gần với trạng thái tự nhiên của phức hợp. Hàm tính điểm của thuật toán docking là phương trình có các tham số cùng với hệ số tuân theo một lý thuyết xác định. Các năng lượng được tính toán là đặc tính năng lượng nội tại của phối tử, năng lượng tự do xoắn và năng lượng giữa các phân tử gồm năng lượng liên kết Van der Walls, năng lượng liên kết hydro, năng lượng từ desolvat và năng lượng tĩnh điện. Mỗi loại tương tác, đều được gán cho 1 miền giá trị, kết quả cuối phản ánh khả năng tương tác mạnh hay yếu của phối tử với enzyme.
Quy trình docking này được sử dụng để sàng lọc các hợp chất Isoflavone được tải về từ CSDL Pubchem sau khi kết quả re-dock 03Q cho thấy tính hợp lý của quy trình. Kết quả protein docking sẽ được lựa chọn dựa trên các tiêu chí sau:
1. Điểm số docking bằng hoặc thấp hơn kết quả chất chứng
2. Cấu dạng có RMSD thấp nhất.
3. Tạo liên kết tốt với các acid amin tại vị trí hoạt động.
Tinh thể 3pp0 được xác định các acid amin quan trọng ở vùng hoạt động bao gồm: LYS753, VAL734, ALA751, GLN799, MET801, LEU852, LEU726, PHE1004, ASP863, ASN850, GLU770, MET774, LEU785, LEU796 [65].
Việc xác định tạo liên kết tốt với các acid amin tại vùng hoạt động là bước quan trọng, nhắm vào vị trí này có thể ức chế hoạt tính của 3pp0 [68].
2.3.2. Nghiên cứu các đặc điểm giống thuốc
Quy tắc Lipinski 5 được sử dụng để so sánh giữa các hợp chất có đặc tính giống thuốc và không giống thuốc sau khi quá trình docking cho thấy khả năng liên kết của phối tử tốt hơn lisinopril đồng kết tinh. Một hợp chất có thể phát triển thành thuốc dùng đường uống nếu không vi phạm quá một trong 4 tiêu chí sau:
- Trọng lượng phân tử: MW < 500 Dalton.
- Số lượng nhóm cho liên kết hydro (Số lượng các nhóm –NH và –OH): HBD < 5.
- Số lượng nhóm nhận liên kết hydro (Bao gồm nguyên tử O và N): HBA < 10.
- Hệ số phân bố octanol/nước: LogP < 5 [52] .
Công cụ trực tuyến pkCSM được sử dụng để đánh giá quy tắc Lipinski
5. Công thức SMILES của các phối tử được lấy từ cơ sở dữ liệu Pubchem
(www.pubchem.ncbi.nlm.nih.gov) và dùng làm dữ liệu đầu vào cho công cụ pkCSM (http://biosig.unimelb.edu.au/pkcsm/prediction).
2.3.3. Nghiên cứu các đặc tính dược động học và độc tính (ADMET)
Sử dụng công cụ trực tuyến pkCSM để dự đoán các đặc tính dược động học và độc tính với dữ liệu đầu vào là công thức SMILES của các hợp chất (Hình 2.3). Kết quả thu được là các thông số về hấp thu (tính tan trong nước, tính thấm màng Caco2, hấp thu ở ruột), phân bố (tính thấm qua hàng rào máu não và hệ thần kinh trung ương...), chuyển hóa (ức chế các enzyme chuyển hóa ở gan), thải trừ ở thận và độc tính (độc tính AMES, độc tính gan...). Một khía cạnh quan trọng trong sự hấp thu chính là tính hấp thu thuốc ở ruột người (HIA) và tính thấm qua màng Caco2. Các hợp chất được cho là hấp thụ kém hoặc hấp thụ vừa hoặc hấp thụ tốt nếu HIA của chúng lần lượt ở trong khoảng 0 đến 20%, 20% đến 70%, và 70% đến 100%. Tính thấm qua màng Caco2 được đánh giá là cao nếu lớn hơn 0,9 (log Papp trong 10 cm/s). Hợp chất có thể thấm qua hàng rào máu não nếu logBB> 0,3 và tương ứng ở hệ thần kinh trung ương (CNS) là trên -2 [73, 74] . Các hợp chất cũng được lựa chọn dựa trên tính không có độc tính của chúng [75].

Hình 2.3. Giao diện công cụ pKCSM.
CHƯƠNG 3. KẾT QUẢ VÀ BÀN LUẬN
3.1. Mô phỏng docking phân tử
3.1.1. Redock 03Q
Trước khi tiến hành docking và sàng lọc các hợp chất, phối tử đồng kết tinh 03Q của tinh thể 3pp0 được tiến hành re-dock lại vào vị trí hoạt động để xác định độ lệch bình phương trung bình gốc (RMSD). Từ đó đánh giá tính phù hợp của quy trình docking. Phối tử đồng kết tinh thu được sau khi re-dock bằng Autodock Vina được so sánh với phối tử đồng kết tinh trước khi re-dock bằng phần mềm Chimera. Giá trị RMSD thu được là 1.076 Å < 1.5 Å, điều này cho thấy quá trình docking, kết quả là phù hợp và đáng tin cậy (Hình 3.1).
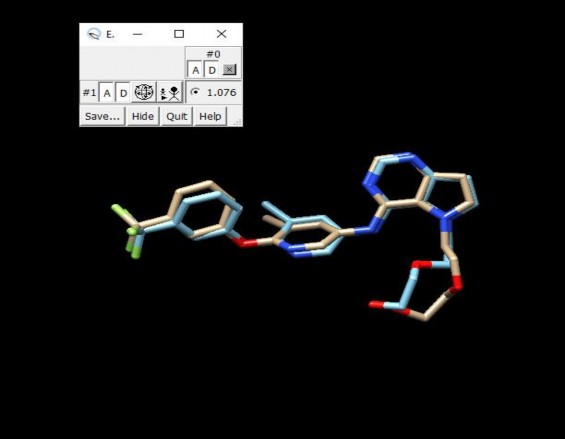
Hình 3.1. RMSD của 03Q đồng kết tinh trước và sau redock
Kết quả năng lượng liên kết khi re-dock 03Q ∆G = -10.7 kCal/mol. Sự tương tác giữa ligand đồng kết tinh và 3pp0 được thể hiện trong Hình 3.2. Phối tử đồng kết tinh cho thấy hình thành liên kết với nhiều acid amin quan trọng ở vùng hoạt động như: liên kết -alkyl với LYS753, VAL 734, ALA751,
MET774, LEU 785, LEU796;-σ với LEU852, LEU726; liên kết hydro với GLN799, MET801, ASP863; - với PHE1004; liên kết Halogen với GLU770; liên kết Van der Waals với ASN850.
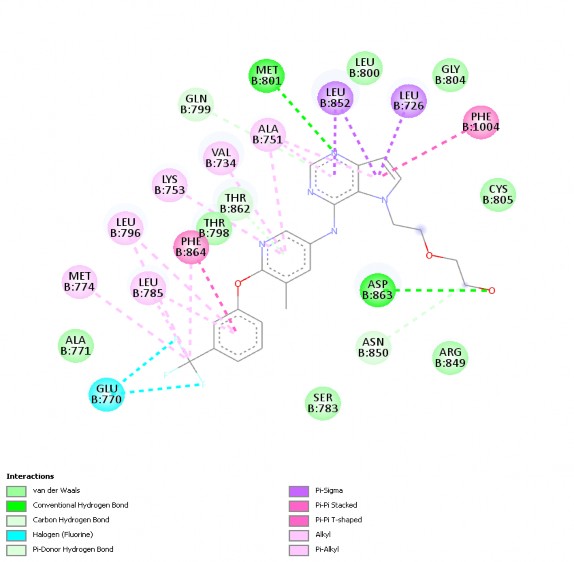
Hình 3.2. Tương tác 2D của ligand đồng kết tinh với 3pp0
Hình 3.3. cho thấy tương tác 2D của ligand đồng kết tinh với 3pp0 sau khi re- dock. Có thể thấy rằng, sau khi re-dock 03Q vẫn liên kết với các nhóm acid amin quan trọng như LYS753, VAL734, ALA751, GLN799, MET801, LEU852, LEU726, PHE1004, GLU770, MET774, LEU785, LEU796 hay
tương tác Van der Waals với ASP863. Sự tương tác của 03Q re-dock chỉ khác với trước khi re-dock là 03Q re-dock không thể hiện được acid amin ASN850. Kết quả này cho thấy mức độ tương tác tốt của ligand với protein khi re-dock.
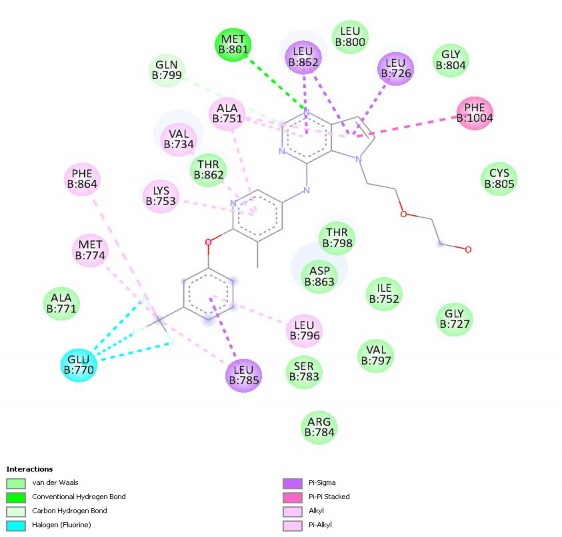
Hình 3.3. Tương tác 2D của ligand đồng kết tinh với 3pp0 sau re-dock.
Như vậy đánh giá chỉ tiêu chính RMSD cùng với đó khả năng tương tác với các acid amin tương đồng giữa cấu trúc 03Q gốc và 03Q re-dock khẳng định quy trình docking là phù hợp để tiếp tục tiến hành quy trình docking 60 hợp chất Isoflavone trong CSDL và hợp chất chứng dương.
3.1.2. Tìm kiếm các chất tiềm năng từ kết quả docking
Sau khi quá trình re-dock chứng minh được sự phù hợp, tiến hành docking 60 hợp chất Isoflavone và các chứng dương vào vị trí hoạt động của protein tại tọa độ (x,y,z) (34, 46, -12) hộp kích thước 30 Å x 30 Å x 30 Å [68]. Năng lượng liên kết ∆G của 60 hợp chất và Trastuzumab được thể hiện ở Bảng 3.1.