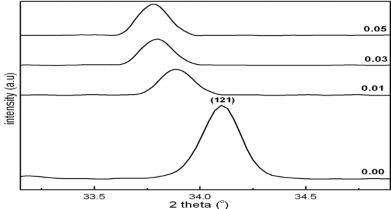
Hình 1.13. Sự dịch của đỉnh nhiễu xạ về phía góc nhỏ quan sát tại đỉnh (121) khi nồng độ Fe tăng.
Có thể nhận thấy rằng khi nồng độ pha tạp tăng lên, vị trí các đỉnh nhiễu xạ bị dịch dần về phía góc nhiễu xạ nhỏ hơn. Điều này có thể được giải thích là do nồng độ Fe tăng lên làm cho các thông số của ô cơ sở cũng tăng lên. Bán kính ion trung bình của Fe4+ là 0.585Å, trong khi đó bán kính trung bình của Mn4+ là 0.53 Å. Điều này làm cho bán kính ion trung bình vị trí B tăng lên và làm cho ô mạng dãn ra.
Nghiên cứu các tán xạ Raman
Vị trí của các đỉnh Raman có sự sai khác không đáng kể khi sử dụng hai bước sóng kích thích khác nhau. Cũng giống như trong nghiên cứu của nhóm tác giả Abrashev [8], trong phổ Raman của gốm CaMnO3 xuất hiện thêm đỉnh Raman ở
vị trí 612cm-1, nhưng có cường độ yếu hơn rất nhiều. Đỉnh Raman này không xuất
hiện trong phổ của các màng mỏng CaMnO3 và có thể được giải thích là do sự có mặt của một pha không tinh khiết, ví dụ CaO dư trong mẫu.
Trong các manganite, sự méo mạng cấu trúc có ảnh hưởng lớn tới cường độ của các đỉnh raman. Các vật liệu có cấu trúc kiểu GdFeO3 như CaMnO3, được mô tả bởi nhóm không gian Pnma (D2h16) có thể được xem như méo mạng trực giao từ cấu
trúc lập phương lý tưởng. Các nguyên tố Ca, Mn, O(1),O(2) lần lượt thuộc nhóm đối xứng Csxz, Ci, Csxz, C1. Các nguyên tố Mn không nằm trong số 24 mode phonon được phép (Raman allowed phonon modes) bao gồm 7 mode đối xứng Ag, 5 mode
B1g, 7 mode B2g và 5 mode B3g. Do sự đối xứng vị trí nguyên tử, 5 trong số 12 tọa
độ nguyên tử được cố định.
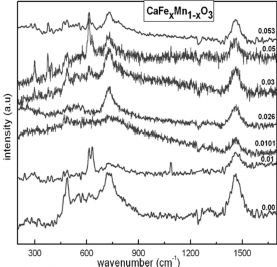
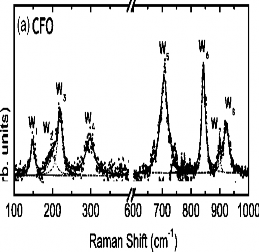
(a) (b)
Hình 1.14. (a)-Phổ tán xạ Raman của CaFeO3 ở 300K (λHe-Ne=632.8 nm) (b)-Phổ tán xạ Raman của CaFexMn1-xO3 ở 300K (λHe-Ne=632.8 nm)
Theo nghiên cứu của các tác giả [8,13] khi pha tạp Fe vào vị trí Mn, trong ô cơ sở của vật liệu sẽ tồn tại sẽ đồng thời các bát diện MnO6 và FeO6. Vật liệu CaFeO3 cũng có cấu trúc GdFeO3, nhóm không gian Pnma và có các mode hoạt động Raman được liệt kê như sau:
ГCaFeO3 = 7Ag + 7B1g + 5B2g + 5B3g. (1.17)
Trong 24 mode hoạt động Raman của CaFeO3 gồm có 16 mode dao động của bát diện FeO6 và 8 mode dao động của ion Ca2+.
Do đó có thể kết luận rằng việc pha tạp Fe làm cho các pha không tinh khiết được tăng cường. Tuy nhiên, chúng ta chưa thể xác định được chính xác pha này do các tính toán lý thuyết hiện có chưa chỉ ra được mode dao động này thuộc về dao động của bát diện FeO6. Cũng tương tự như vậy, có thể kết luận rằng đỉnh Raman
711 cm-1 trong hệ gốc tương ứng biến dạng giãn của bát diện MnO6 có cường độ
giảm dần khi nồng độ pha tạp tăng lên. Trong khi đó vị trí số sóng 728 cm-1 tương
ứng với biến dạng giãn bất đối xứng của bát diện BO6 lại được tăng cường khi nồng
độ Fe tăng lên. Điều này có thể được giải thích là do sự thay thế của Fe vào vị trí Mn làm bát diện BO6 bị giãn bất đối xứng. Tuy nhiên sự sai khác về bán kính ion của Mn và Fe nhỏ, nên đỉnh Raman tương ứng biến dạng giãn của bát diện BO6 có sự dịch không đáng kể.
Phổ hấp thụ hồng ngoại
Các phép đo phổ hồng ngoại là một công cụ hiệu quả để nghiên cứu các hệ vật liệu có liên kết mạnh giữa điện tử và mạng tinh thể.
Ngoài ra cũng có sự dịch các đỉnh hấp thụ về phía đỏ (red-shift) khi nồng độ pha tạp tăng lên. Các ước đoán độ rộng vùng cấm cũng chỉ ra khi nồng độ pha tạp tăng lên, độ rộng vùng cấm có xu hướng giảm đi. Điều này có thể được giải thích là do khi nồng độ Fe tăng lên làm cho mật độ điện tử 3d trong tinh thể tăng lên. Điều này chủ yếu có nguồn gốc từ trạng thái hóa trị 2+ của Fe. Khác với Mn, một phần ion Fe tồn tại trong trạng thái hóa trị 2+, dẫn đến một phần dư điện tử lớp 3d được bơm lên vùng dẫn. Sự dịch chuyển đỏ có thể là dấu hiệu của quá trình này.
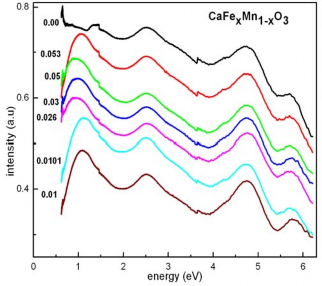
Hình 1.15. Phổ hấp thụ của các mẫu gốm CaFexMn1-xO3 đo tại nhiệt độ phòng
chỉ ra phổ hấp thụ của các mẫu gốm CaFexMn1-xO3 đo tại nhiệt độ phòng. Từ phổ hấp thụ của các mẫu có thể quan sát thấy có 4 đỉnh hấp thụ tương ứng năng lượng cỡ khoảng 6.7, 4.7, 2.5 eV. Riêng đối với các mẫu pha tạp, xuất hiện thêm một đỉnh hấp thụ rò nét tương ứng năng lượng khoảng 1.2 eV.
CHƯƠNG 2
CÁC PHƯƠNG PHÁP THỰC NGHIỆM
2.1. Phát tán siêu âm để tạo hạt nano trong dung môi hữu cơ
Các dung dịch hạt nano được tạo ra theo quy trình chung như sau. Trước hết, các hóa chất được sử dụng phải có độ tinh khiết cao > 99.9 %. Nước tinh khiết cũng phải là nước cất RO hai lần, có điện trở suất lớn hơn 108 Ωcm. Các chất được sử dụng bao gồm: Fe3O4, nước cất RO hai lần, chất hoạt hoá bề mặt Span-80, aceton. Để tạo được các dung dịch cần nghiên cứu, chúng tôi đã thực hiện ba bước chính sau đây.
Bước thứ nhất, nghiền các bột Fe3O4 có khối lượng 0.6 g trong nước tinh khiết để được các hạt ở dạng kích thước nano, sau đó bổ sung thêm 30 ml nước để tạo được dung dịch Fe3O4 có nồng độ là 2%. Tiếp theo khuấy từ trong 1h và siêu âm dung dịch trong thời gian 20 phút (nguồn siêu âm công suất 30 W). Sau khi siêu âm các dung dịch này được quay li tâm với tốc độ 3.500 vòng/phút trong thời gian 30 phút. Lọc phần cặn, lấy phần trong bên trên tách ra 10 ml các dung dịch trong suốt.
H2O | |||
Có thể bạn quan tâm!
-

 Tính chất quang của chất keo Fe3O4 chức năng hóa bề mặt trong từ trường - 1
Tính chất quang của chất keo Fe3O4 chức năng hóa bề mặt trong từ trường - 1 -
 Tính chất quang của chất keo Fe3O4 chức năng hóa bề mặt trong từ trường - 2
Tính chất quang của chất keo Fe3O4 chức năng hóa bề mặt trong từ trường - 2 -
 Tính Siêu Thuận Từ Của Hạt Nano Từ; (A) Moment Từ Hướng Theo Phương Trục Dễ Của Hạt T< T B . ; (B) Moment Từ Hướng Theo Từ Trường Ngoài T > T B
Tính Siêu Thuận Từ Của Hạt Nano Từ; (A) Moment Từ Hướng Theo Phương Trục Dễ Của Hạt T< T B . ; (B) Moment Từ Hướng Theo Từ Trường Ngoài T > T B -
 Sơ Đồ Chi Tiết Máy Đơn Sắc Ms257 Và Máy Quang Phổ Cùng Các Phụ Kiện
Sơ Đồ Chi Tiết Máy Đơn Sắc Ms257 Và Máy Quang Phổ Cùng Các Phụ Kiện -
 Tính chất quang của chất keo Fe3O4 chức năng hóa bề mặt trong từ trường - 6
Tính chất quang của chất keo Fe3O4 chức năng hóa bề mặt trong từ trường - 6 -
 Tính chất quang của chất keo Fe3O4 chức năng hóa bề mặt trong từ trường - 7
Tính chất quang của chất keo Fe3O4 chức năng hóa bề mặt trong từ trường - 7
Xem toàn bộ 65 trang tài liệu này.
- Khuấy từ
- Rung siêu âm
Dung dịch Fe3O4 (Màu trắng đục)
- Quay ly tâm
- Lọc bỏ cặn, lấy phần trong
Dung dịch Fe3O4
(Trong suốt)
Hình 2.1. Sơ đồ chế tạo dung dịch Fe3O4
Bước thứ hai, hoà tan 40 ml aceton có khối lượng 31.7 g với 0.15 ml span có khối lượng 0.15 g để được dung dịch span và aceton 0.5 %, mặc dù chúng đã hoà tan vào nhau nhưng vẫn cần phải siêu âm dung dịch này trong thời gian 15 phút, tỷ lệ pha giữa aceton với span quyết định nhiều đến chất lượng mẫu tạo ra.
Span-80 | |||
- Rung siêu âm
Dung dịch span và aceton 0,5%
Hình 2.2. Sơ đồ chế tạo dung dịch span và aceton
Dung dịch nano sắt từ CaFexMn1-xO3 cũng được chế tạo bằng cách tương tự.
Tỷ lệ pha trộn thể tích | |
Mẫu 1 Fe3O4 nồng độ 1-4 | 1ml dung dịch Fe3O4 4ml dung dịch Span và Aceton |
Mẫu 2 Fe3O4 nồng độ 1-2 | 1ml dung dịch Fe3O4 2ml dung dịch Span và Aceton |
Mẫu 3 CaFexMn1-xO3 nồng độ 1-8 | 1ml dung dịch CaFexMn1-xO3 8ml dung dịch Span và Aceton |
Mẫu 4 CaFexMn1-xO3 nồng độ 1-16 | 1ml dung dịch CaFexMn1-xO3 16ml dung dịch Span và Aceton |
Các mẫu chế tạo phục vụ thực nghiệm, nồng độ pha tạp của Fe trong các mẫu CaFexMn1-xO3 là 0.025
Bước cuối cùng là lần lượt pha dung dịch aceton và span 0,5% với dung dịch Fe3O4 và CaFexMn1-xO3 theo những tỷ lệ thể tích khác nhau. Kết quả được thể hiện trên Bảng 2.1.
2.2. Các phương pháp nghiên cứu
2.2.1. Phổ hấp thụ UV-Vis
Cách tử | Khe 1 | Nguồn sáng UV Nguồn sáng VIS | Gương 1 Io | ||
Mẫu so sánh Gương 2 | Thấu kính | Đầu thu1 I | Bộ xử lí | ||
Gương 3 | Mẫu đo | Thấu kính | Đầu thu2 |
Hình 2.3. Sơ đồ khối của thiết bị UV-Vis Agilent 8453
Để xác định phổ hấp thụ của một màng mỏng, một nguồn sáng với dải phổ rộng được chiếu xuyên qua màng, cường độ của ánh sáng đi ra được một đầu thi ghi lại từng giá trị ứng với từng bước sóng. Giá trị cường độ này được so sánh với giá trị cường độ ánh sáng ban đầu sau khi tính đến khi sự hấp thụ của vật liệu đế được gọi là mật độ quang học. Mật độ quang học là đại lượng tương đối và có giá trị là:
D() ln( I0 )
I
(2.1)
Trong đó I0 và I tương ứng là cường độ ánh sáng tới và ánh sáng truyền qua với mỗi bước sóng λ. Khi ánh sáng truyền qua vật liệu trong suốt, nếu cường độ ánh sáng tới là I0 thì cường độ I được truyền qua mặt sau của mẫu tuân theo định luật Buger- Lamber- Beer:
I I0 (1R) exp(d )
(2.2)
Trong đó α gọi là hệ số hấp thụ của vật liệu. R là hệ số phản xạ của vật liệu.
Hệ số hấp thụ của vật liệu được xác định theo công thức:
() 1 ln( I )
(2.3)
d I0 (1R)
Nếu sự phản xạ trên bề mặt mẫu là không đáng kể thì hệ số hấp thụ được tính theo công thức:
() 1 ln( I ) D
(2.4)
d I0 d
Đối với bán dẫn vùng cấm thẳng, độ rộng vùng cấm của vật liệu có thể xác
định theo hệ số hấp thụ của vật liệu theo công thức sau:
g
[(h)]2 C(hE )
(2.5)
Từ công thức trên, độ rộng vùng cấm của vật liệu có thể xác định bằng cách
vẽ đường biểu diễn đại lượng
[(h)]2 theo năng lượng, sau đó dựng tiếp tuyến
với đường nói trên và giao điểm của tiếp tuyến này với trục năng lượng cho giá trị độ rộng vùng cấm.
2.2.2. Phổ huỳnh quang
Phổ huỳnh quang thể hiện phân bố năng lượng vùng hóa trị và phổ hấp thụ thể hiện phân bố năng lượng vùng dẫn trên mức Fermi. Thông thường chúng ta thu được phổ huỳnh quang do vật liệu bức xạ ra dưới tác dụng kích thích của ánh sáng tử ngoại. Cũng như phổ hấp thụ, phổ huỳnh quang phụ thuộc vào thành phần và cấu trúc của các tâm bức xạ và các tác nhân bên ngoài. Phổ huỳnh quang có một số đặc điểm sau:
- Phổ huỳnh quang bao giờ cũng có tần số bé hơn tần số của ánh sáng kích thích. Tần số huỳnh quang trong trường hợp này gọi là tần số Stocke. Trong những trường hợp đặc biệt, do cấu trúc của các mức năng lượng hoặc do phương pháp kích
thích khác nhau có thể xảy ra trường hợp va chạm giữa hai phân tử kích thích và điện tử để lên mức năng lượng cao hơn rồi từ đó chuyển về trạng thái cơ bản bức xạ ra tần số lớn hơn tần số của ánh sáng kích thích. Tần số này gọi là tần số đối Stocke.
’
Máy thu phổ
Mẫu
L
Hình 2.4. Cách bố trí thu phổ huỳnh quang
- Phổ huỳnh quang không phụ thuộc vào phổ của ánh sáng kích thích.
- Phổ huỳnh quang phụ thuộc rất nhiều vào những tạp chất nằm trong mẫu. Cách xác định phổ huỳnh quang: chiếu bức xạ đơn sắc của đèn thủy ngân,
đèn xênôn hoặc laser kích thích chất huỳnh quang và đo I = f(). Gọi Bhq(1) là độ chói của ánh sáng huỳnh quang của mẫu nghiên cứu ứng với tần số 1. Bng(1) là độ chói của ánh sáng của nguồn chuẩn ứng với tần số 1. a(1) là hệ số tỷ lệ của hai độ chói. Khi đó ta có:
Bhq(1) = a(1) Bng(1) (2.6) Tương tự đối với tần số 2, i và max ta có:
Bhq(2) = a(2) Bng(2) (2.7)
Bhq(i) = a(i) Bng(i) (2.8)