By-products of the reaction include: curcumin raw material, mono -O-( 2-hydroxyethyl)curcumin compound ( PH6 ) and possibly tri-(2-hydroxyethyl)curcumin ( PH6c, PH6d ) ( Figure 4.3 ) . To obtain high yield of PH7 product , it is necessary to pay attention to control the molar ratio of curcumin: 2-bromoethanol for this reaction. In addition, as with the reaction to create monohydroxyethyl derivatives, the reaction conditions to create PH7 must be dry to avoid damaging the bromethanol agent and generating other oxidizing impurities.
4.1.4. Discussion on the reaction to form monoester derivatives of glutarate (PH8) and succinate (PH9 and PH10)
4.1.4.1. On the synthesis of mono-O-(2-(succinyloxy)ethyl)curcumin derivative (PH9)
Maybe you are interested!
-

 Discussion on Causes, Limitations and Existing Problems of Short-Term Loan Service Quality Khdn
Discussion on Causes, Limitations and Existing Problems of Short-Term Loan Service Quality Khdn -
 Discussion of Quantitative Research Results on the Impact of Control Variables on Financial Performance
Discussion of Quantitative Research Results on the Impact of Control Variables on Financial Performance -
 Main Discussion Issues System
Main Discussion Issues System -
 Phenomenon that changes reaction rate - 2
Phenomenon that changes reaction rate - 2 -
 Discussion of Research Results of the First Model
Discussion of Research Results of the First Model

The acylation reaction of PH6 using succinic anhydride as a catalyst with pyridine as a promoter can occur according to two mechanisms (a) and (b) shown in the following diagram ( Figure 4.5 ) [12], [66]:
Figure 4.5. Schematic diagram of the acylation reaction mechanism to create PH9
In the PH9 molecular structure, there is still 1 free –COOH group and 1 ester group that can be hydrolyzed, so the reaction needs to be carried out at a cold temperature of 5–10 °C and needs to be processed quickly to avoid hydrolysis of the ester bond. To limit the product from entering the aqueous phase and remove ionic impurities, we use washing with saturated NaCl solution, then washing again with water.
4.1.4.2. On the synthesis of sodium salt of mono-O-(2-(succinyloxy)ethyl)curcumin (PH10)
NaHCO 3 is used to selectively create PH10 because NaHCO 3 does not create phenolate salts as by-products and NaHCO 3 is a weak alkali (pH of saturated solution is 8.4), so it limits hydrolysis of ester bonds compared to stronger alkalis such as NaOH and Na 2 CO 3 .
To select the reaction solvent, we based on the solubility of NaHCO 3 (at 22 °C): very soluble in water, insoluble in ethanol, slightly soluble in acetone (0.02%), soluble in methanol (2.13%) and of PH9 (slightly soluble in methanol and acetone). Accordingly , acetone has an advantage over methanol: acetone dissolves less NaHCO 3 than methanol, limiting the amount of residual hydrocarbonate in the solution after filtration. In addition, acetone also helps the obtained product to be more stable, while methanol has flexible hydrogen that can hydrolyze/alcoholize ester bonds in alkaline environments.
Residual alkali affects the stability of the product during storage, so the amount of NaHCO 3 used in the reaction must be sufficient to completely convert the PH9 acid form into the PH10 salt form and minimize the amount of excess NaHCO 3 , so it is necessary to strictly control the molar ratio of NaHCO 3 when performing the reaction.
During the experiment, PH10 was synthesized through two methods: creating PH10 through the PH9 separation stage (method A) and creating PH10 salt from PH6 without going through the PH9 separation stage (method B). The results showed that method B was easier to implement, saved more time, and had higher process efficiency (67.38% compared to 52.48%) compared to method A because PH9 was partially lost during the precipitation washing process with methanol. Therefore, method B was chosen as a direction for one-pot synthesis of mono- O -(2-(succinyloxy)ethyl)curcumin sodium salt ( PH10 ).
4.1.4.3. On the synthesis of mono-O-(2-(glutaryloxy)ethyl)curcumin derivative (PH8)
The synthesis of PH8 is an acylation reaction of PH6 using glutaric anhydride as a pyridine catalyst, which is similar to the synthesis of PH9 in terms of mechanism, reaction conditions and possible by-products. However, because glutaric anhydride is a weaker acylating agent than succinic anhydride, the process uses a higher molar ratio of anhydride and a longer reaction time than the synthesis of PH9 . Experiments also show that using the extraction method (cold HCl and saturated CuSO 4 ) significantly decomposes the product and almost does not yield PH8 as expected. Therefore, we used column chromatography for purification.
4.1.5. Discussion on the reaction to form glutarate diester derivatives (PH14) and succinate (PH15)
4.1.5.1. On the synthesis of di-O-(2-(glutaryloxy)ethyl)curcumin (PH14)
In 2018, Muangnoi and colleagues performed a direct acylation reaction of curcumin with glutaric anhydride to produce soluble curcumin derivatives [113]. The authors used dichloromethane as solvent and triethylamine as catalyst, at a temperature of 40 °C.
(reflux) for 2 hours for the diacylated product with an 85% yield. However, when we used this condition to glutarylate curcumin and PH7 , the reaction was almost poor and the product could not be purified, especially with curcumin. Therefore, we replaced dichloromethane with chloroform solvent to increase the temperature (reflux) and shorten the reaction time and only performed with PH7 raw material to create PH14 . In particular, chloroform needs to be purified immediately before performing acylation for good efficiency.
4.1.5.2. On the synthesis of di-O-(2-(succinyloxy)ethyl)curcumin (PH15)
The acylation reaction of PH7 to form PH15 using succinic anhydride as a catalyst with triethylamine occurred according to the same mechanism as the reaction to form PH9 ( Figure 4.5 ) and was carried out according to the method of Muangnoi et al. (2018) [113], similar to the synthesis process of PH14 . Compared with the synthesis of PH14 (glutaric anhydride), the synthesis reaction of PH15 used a stronger acylating agent, less amount of agent used and shorter reaction time.
Thin layer chromatography showed that the reaction did not produce a single product, but a main spot and several secondary spots appeared. These secondary spots were predicted to be raw material PH7 , mono ester impurity PH15a and tri ester impurity PH15b ( Figure 4.6 ). When processing the reaction, we found isopropanol as the solvent to remove these impurities.
Figure 4.6. Impurity generation possibilities of PH15 synthesis reaction
During the esterification process, it is necessary to control the concentration of the anhydride agent. Dilute concentrations (solvent amount > 30 mL) lead to a longer reaction time, while not increasing the reaction yield. Concentrated concentrations of the agent (solvent amount < 10
mL) increases the possibility of creating PH15b impurities . We chose a reasonable amount of solvent as 20 mL. It should be noted that PH15 has intramolecular ester and COOH bonds, so the reaction mass needs to be processed under cold conditions. During the rotary distillation stage, the solvent needs to be distilled at low temperature (30–35 °C) to avoid hydrolysis of the ester bond.
4.1.6. Discussion on the reaction to form phosphate derivatives PH11 and PH12
4.1.6.1. On the synthesis reaction of 2-(curcumin-O-yl)ethyl dihydrophosphate (PH11)
Phosphation reaction to create PH11 can be performed by several methods such as: Using phosphoric acid or phosphoryl chloride. Experiments show that using phosphoric acid gives low yield (12.71%). We tested phosphoryl chloride according to the method of


T. Sowa [162] obtained better results (67.74%). Phosphory chloride combined with pyridine and water has some advantages such as: selectivity to primary alcohol, relatively high efficiency, suitable for laboratory conditions, so it was chosen in this reaction. The mechanism of the reaction can occur as shown in the following figure [162]:
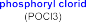
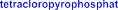
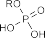

Figure 4.7. Mechanism of phosphorylation reaction using POCl 3 agent [162]
Phosphorylate chloride combines with pyridine and water at a molar ratio of 2:2:1 to form the intermediate tetrachloropyrophosphate (P 2 O 3 Cl 4 ) which is a selective phosphatizing agent (only forming monophosphate on the primary alcohol OH of PH11 , without affecting the phenol and enol OH of the curcumin structural framework).
The surveys show that the optimal molar ratio POCl 3 : PH6 = 15 : 1, different from the referenced document (molar ratio POCl 3 : PH6 = 5 : 1). The remaining parameters are similar to the reference document: temperature 0–2 °C, solvent acetonitrile, reaction time 4 hours. Cold temperature helps limit the hydrolysis of POCl 3 and P 2 O 3 Cl 4 into H 3 PO 4 . The amount of water added to the reaction block also needs to be controlled at the following ratio: POCl 3
: water : pyridine = 2: 1: 2.
During the reaction, excess PH6 impurities are removed by washing with diethyl ether; and water-soluble impurities (pyridine hydrochloride salt, inorganic phosphate) are also removed when extracting the product with ethyl acetate. Experiments show that the crude acid product PH11 is unstable and easily hydrolyzed to PH6 due to the presence of ester and intramolecular acid groups. Adjusting the pH to 1-2 helps neutralize free phosphoric acid (generated during the reaction), thereby limiting the hydrolysis of PH11 product in the following steps.
4.1.6.2. On the reaction to form disodium salt 2-(curcumin-O-yl)ethyl phosphate (PH12)
The reaction to create PH12 is a reaction to create acid hydrophosphate and alkaline carbonate salts with quite good yield (95.47%). When treating the reaction solution (pH 9) with ether, almost no trace of PH11 material was observed in the second and third extracts. Experiments show that PH12 is almost insoluble in ether.
4.1.7. Discussion on the reaction to form PH13 sulfate derivatives
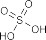
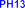
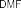
The reaction was carried out according to the method of author Mumma (1966) [115]: synthesizing sulfate ester via DCC intermediate by attaching sulfate group through oxyethyl bridge of intermediate PH7 to form PH13. The reaction mechanism of sulfation of intermediate PH7 is explained in document [18] shown in figure 4.8 below. The SO bond of sulfuric acid is activated by DCC via intermediate A, then reacts with OH group in PH7 molecule to form esterification product.
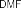
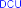
Figure 4.8. PH7 sulfation reaction mechanism mediated by DCC [18]
The order of addition of the reactants is an important factor leading to the formation of the sulfate product. The reaction we carried out was in the order: (1) DCC, (2) nucleophil (alcohol PH7 ), (3) H 2 SO 4. If the reaction were carried out in the order: (1) H 2 SO 4 , (2) nucleophil , (3) DCC, the nucleophil would decompose before DCC was added. In addition, if the reaction order was: (1) DCC, (2) H 2 SO 4 , (3)
nucleophile, the sulfate product is also not formed because DCC and H 2 SO 4 react directly with each other in DMF solvent to form dicyclohexylure [68]. DCC is hygroscopic and causes strong allergies when in contact with the skin, so when performing the reaction, it is necessary to operate quickly and carefully. The reaction needs to be maintained at low temperature throughout the process to avoid sulfation of DMF and avoid creating other by-products [115]. In addition, PH7 has 2 alcohol OH groups, so it can form non-selective sulfation products on 1 or both of these groups ( Figure 4.9 ). We have chosen the molar ratio of the reactants (nucleophine: DCC: H 2 SO 4 = 1: 5: 1) and dilute concentration to control, giving priority to creating monosulfation products.
Figure 4.9. Possible products in the PH13 synthesis reaction
During the treatment process, the aqueous phase was washed with dichloromethane and ethyl acetate to remove most of the DMF as well as the raw material impurities. The DCU impurities were precipitated in methanol and filtered away. The disulfate by-product was poorly soluble in isopropanol, so we used isopropanol for purification.
4.1.8. Discussion on PH16 conjugation reaction
On the N-Boc-valine synthesis reaction:
Hybridization of curcumin and amino acids has been studied by some authors to improve solubility, in which it is possible to use an ester bridge directly with the phenolic OH of the curcumin skeleton. The resulting amino group (-NH 2 ) has the ability to form salts at acidic pH, thereby improving the solubility of the derivative [128]. To synthesize PH16 from PH7 derivative and L -valine via an ester bridge, it is necessary to protect the -NH 2 group of valine. Some NH 2 protecting groups that can be used are 9-fluorenylmethoxycarbonyl (Fmoc), tert -butoxycarbonyl (Boc), trityl (Trt), allyloxycarbonyl (Alloc) [71]. Of which, Fmoc and Boc are the most commonly used due to their easy attachment and selective removal with acid solutions.
trifluoroacetic acid (TFA) 20-50% in dichloromethane or 5M hydrochloric acid solution, 85% phosphoric acid solution. On that basis, we chose the Boc group because it is available in the laboratory (Boc 2 O) and easy to choose the removal conditions later. The reaction mechanism for protecting valine with Boc is shown in Figure 4.10 .

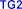
Figure 4.10. Mechanism of N-Boc-valine synthesis reaction
The reaction was carried out based on the literature of Mishra et al. (2014) [106], with some changes in reaction time (96 hours compared to 12 hours in the literature) and reaction temperature (heating at ~50 °C instead of room temperature).
About the total reaction TG3:
The reaction was carried out according to the document of KS Parvathy et al. [128]. We found that it is necessary to use more dicyclohexylcarbodiimide (DCC) than the document for the reaction to occur completely. The dicyclohexylcarbodiimide agent and the auxiliary amine 4-dimethylaminopyridine are hygroscopic substances, so it is necessary to carry out in anhydrous solvent to achieve good results. In addition, when cooling, it is also necessary to pay attention to keeping the environment dry, avoiding condensation of water vapor that can damage the DCC agent. Temperature does not significantly affect the efficiency of the reaction. Dicyclohexylurea (DCU) impurities easily precipitate in the reaction mass, which we removed by filtration and treatment with ethyl acetate.
About PH16 synthesis reaction
Hydrolysis of the –Boc group can use some acidic conditions such as: trifluroacetic acid in dichloromethane, HCl in ethyl acetate, H 2 SO 4 in tert -butyl methyl ester, phosphoric acid in tetrahydrofuran or Lewis acids such as TiCl 4 , SnCl 4 , AlCl 3
... TG3 has two hydrolyzable bonds, amide and ester. Therefore, it is necessary to choose a selective hydrolyzing agent –Boc (amide) that does not hydrolyze the ester function. Based on the document of FS Gibson (1994) [54], we choose the following conditions:
+ Hydrolyzing agent HCl k in methanol (document is HCl k in ethyl acetate).
+ Reaction time longer than 8-12 hours (document 2 hours).
+ The reaction temperature is ~ 50 °C (document is room temperature).
HCl k in methanol has the advantage of being easy to distill after the reaction is complete. The excess HCl is treated with saturated NaHCO 3 solution at pH = 8-9. The product is extracted with ethyl acetate, then purified by column chromatography.
4.2. DISCUSSION ON STRUCTURAL DETERMINATION OF DERIVATIVES

4.2.1. Discussion on the structures of PH1 and PH2 derivatives
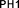
Figure 4.11. Structures of PH1 , PH2 and proton signal values on 1 H-NMR spectrum
According to the document published in 2002 [119], the product obtained from the alkylation reaction of curcumin with methyl chloroacetate is a di-alkylated derivative on the 2 -OH phenol groups of curcumin. However, the results of our spectral data analysis showed that the product obtained in this study is a tetra-alkylated derivative ( PH1 ). Specifically, the ESI-MS spectrum of PH1 gives the molecular ion peaks [M+Na] + ( m/z = 679.2) and [MH] - ( m/z = 655.2) consistent with the molecular mass of curcumin substituted by 4 methoxycarbonylmethyl groups of 656.64 uC, corresponding to the molecular formula C 33 H 36 O 14 [ Appendix 2 ].
The characteristic bonds for the curcumin skeleton and these substituents are also shown in the IR spectrum of PH1 , which are: aromatic CH and conjugated double bonds (with ν̅ max = 3010 cm - 1 , weak intensity), saturated CH (2951 and 2866 cm - 1 , weak), C=O ketone (1665 cm - 1 , strong), aromatic C=C (1587 and 1509 cm - 1 , medium), CO (1259 and 1189 cm - 1 , strong); in which there is no tautomerization of the β-dicetone structure because the proton at C-4 has been completely substituted. In addition, there is also a characteristic absorption peak of the C=O ester group (1741 cm - 1 , strong), proving that the esterification reaction of -OH phenol has occurred [ Appendix 1 ].
On the 1 H-NMR spectrum of PH1, no singlet signals of the groups were observed.
The free phenolic -OH (with Ł ~ 9.64 ppm) as well as that of the proton at C-4 (with Ł ~ 3.90 ppm - for the keto form or Ł ~ 6.0 ppm - for the enol form) - also demonstrate the positions