Based on previous publications, 60 isoflavones were identified as HER2 inhibitors [58-65]. The compounds were downloaded from the Pubchem database (http://pubchem.ncbi.nlm.nih.gov/). This is the world's largest freely accessible database of chemical compounds, maintained by the National Center for Biotechnology Information, part of the US National Library of Medicine.
2.1.3. Evidence structure
Trastuzumab, Lapatinib, Tucatinib are anti-cancer drugs, approved by the FDA (US Food and Drug Administration) in the treatment of HER2-positive breast cancer [35,44,66]. Compare the docking scores of Isoflavone compounds with these substances to evaluate and screen for HER2 enzyme inhibition ability.
2.1.4. Equipment and software
Device used: Desktop computer generation core i5-10500 and GTX 1660.
Maybe you are interested!
-

 Diagnosis of laryngeal cancer by indirect rigid endoscopy and flexible tube biopsy - 1
Diagnosis of laryngeal cancer by indirect rigid endoscopy and flexible tube biopsy - 1 -
 Let Li And Et Al (2002), "prevalence Of Breast - Feeding And Its Correlates In Ho Chi Minh City Vietnam", Pediatrics International (44), Pp.47-54.
Let Li And Et Al (2002), "prevalence Of Breast - Feeding And Its Correlates In Ho Chi Minh City Vietnam", Pediatrics International (44), Pp.47-54. -
 Study on clinical features, pathology and treatment results of non-epithelial gastrointestinal cancer at Viet Duc Hospital - 17
Study on clinical features, pathology and treatment results of non-epithelial gastrointestinal cancer at Viet Duc Hospital - 17 -
 Study on chemical composition and evaluation of anti-cancer effect of stem and leaves of Stephania dielsiana YC Wu - 1
Study on chemical composition and evaluation of anti-cancer effect of stem and leaves of Stephania dielsiana YC Wu - 1 -
 Computer simulation, assembly and quotation software - 2
Computer simulation, assembly and quotation software - 2
Windows 11 operating system.
Software: The following software and tools were used in the study:
- MGL tools 1.5.6 ( http://mgltools.scripps.edu/)
- Autodock Vina 4.2 ( http://vina.scripps.edu/)
- UCSF Chimera 1.15 ( https://www.cgl.ucsf.edu/)
- Avogadro 1.2.0 ( http://avogadro.cc/)
- Discovery Studio 2021 Client (https://discover.3ds.com/)
- Microsoft Office 2016.
- Microsoft Excel 2016.
2.2. Research content
Step 1: Screening of isoflavone compounds downloaded from Pubchem database for their ability to inhibit HER2 enzyme using molecular docking method. Comparing the energy with reference substances to select ligand-protein complexes with suitable binding energy.
Step 2: Study the drug-like properties of the compounds with the best molecular docking screening results.
2.3. Research method
2.3.1. Protein docking simulation
2.3.1.1. Re-dock 03Q
Re-docking is an important step to validate the docking process. In the protein 3pp0, there is a co-crystallized ligand, 03Q, Figure 2.2 shows the 2D structure of 03Q. This co-crystallized ligand is re-docked into the active site of the protein. If the root mean square deviation (RMSD) value is less than or equal to 1.5 Å, the suitability of the docking process can be concluded [67]. Steps to perform re-docking:
Protein preparation : The crystal structure of 3pp0 was downloaded from Protein Data Bank. Water molecules were removed and ligands were co-crystallized using Discovery Studio software. Next, Autodock Tools 1.5.6 was used to add hydrogens, optimize polar hydrogens, and attach the Kollman force field. The active site of the protein was defined in a grid box of size 30 Å x 30 Å x 30 Å, with the center (x,y,z) at (34, 46, -12) [68]. The protein was then saved in pdbqt format to prepare for docking.
Preparation of co-crystallized ligand : Open protein 3pp0 with Discovery Studio software. Remove water, protein and keep only other molecules. Save the file with .pdb extension. Convert ligand to .pdbqt extension with Autodock Tools software.
Figure 2.2. 2D structure of cocrystallized ligand 03Q.
Re-dock : The built pdbqt files of co-crystallized ligand and protein were docked using Autodock Vina. The number of iterations for the program was 8. Calculate the RMSD value between 03Q when separated and after re-docking using Chimera software.
2.3.1.2. Protein docking simulation with 60 ligands and controls
Protein preparation: the protein in the docking section was also de-watered, ligand co-crystallized using Discovery Studio; hydrogen was added, polar hydrogens were optimized and Kollman force field was attached and pdbqt file was built using Autodock Tools as done in re-dock.
Preparation of ligands and controls: The 3D structures of these compounds were obtained from the PubChem database in sdf format and then converted to pdb format using Chimera software [69,70]. Next, the ligands were optimized using Avogadro software using the Conjugate Gradients method and then converted to pdbqt format using Autodock Tools 1.5.6 software [71,72].
Docking: The constructed pdbqt files of the ligand and protein were docked using Autodock Vina. The software was used to find the best binding conformation using binding free energy assessments.
∆G and the number of physical interactions. The docking results are evaluated using three criteria: docking score, interaction ability, and RMSD (root mean square deviation).
In docking simulations, the lower the binding energy is considered to be, the closer it is to the native state of the complex. The scoring function of the docking algorithm is an equation whose parameters and coefficients follow a certain theory. The energies calculated are the intrinsic energy properties of the ligand, torsional free energy, and intermolecular energies including Van der Walls binding energy, hydrogen bond energy, desolvation energy, and electrostatic energy. Each type of interaction is assigned a domain value, the final result reflecting the strong or weak interaction ability of the ligand with the enzyme.
This docking procedure was used to screen Isoflavone compounds downloaded from Pubchem database after the 03Q re-dock results showed the validity of the procedure. The protein docking results will be selected based on the following criteria:
1. Docking score equal to or lower than the qualitative result
2. The conformation with the lowest RMSD.
3. Create good bonds with amino acids at the active site.
The 3pp0 crystal identified important amino acids in the active site including: LYS753, VAL734, ALA751, GLN799, MET801, LEU852, LEU726, PHE1004, ASP863, ASN850, GLU770, MET774, LEU785, LEU796 [65].
Identifying the best binding site for amino acids in the active site is a key step, targeting this site can inhibit the activity of 3pp0 [68].
2.3.2. Research on drug-like characteristics
Lipinski's rule of 5 is used to compare drug-like and non-drug-like compounds after docking has shown that the ligand binds better than co-crystallized lisinopril. A compound can be developed into an oral drug if it does not violate more than one of the following four criteria:
- Molecular weight: MW < 500 Dalton.
- Number of hydrogen bond groups (Number of –NH and –OH groups): HBD < 5.
- Number of hydrogen bond accepting groups (Including O and N atoms): HBA < 10.
- Octanol/water partition coefficient: LogP < 5 [52] .
The online tool pkCSM is used to evaluate the Lipinski rule.
5. SMILES formulas of ligands are taken from Pubchem database
(www.pubchem.ncbi.nlm.nih.gov) and used as input data for the pkCSM tool (http://biosig.unimelb.edu.au/pkcsm/prediction).
2.3.3. Study of pharmacokinetic and toxicological properties (ADMET)
The online tool pkCSM was used to predict pharmacokinetic and toxicological properties with input data as SMILES formulas of compounds ( Figure 2.3 ). The results were parameters of absorption (water solubility, Caco2 membrane permeability, intestinal absorption), distribution (permeability through the blood-brain barrier and central nervous system, etc.), metabolism (inhibition of liver metabolizing enzymes), renal excretion, and toxicity (AMES toxicity, hepatotoxicity, etc.). An important aspect of absorption is the human intestinal absorption (HIA) and the permeability through Caco2 membrane. Compounds were considered poorly absorbed, moderately absorbed, or well absorbed if their HIA was in the range of 0 to 20%, 20% to 70%, and 70% to 100%, respectively. The permeability through Caco2 membrane was considered high if it was greater than 0.9 (log Papp in 10 cm/s). Compounds can penetrate the blood-brain barrier if logBB>0.3 and the corresponding central nervous system (CNS) is above -2 [73, 74]. Compounds are also selected based on their non-toxicity [75].

Figure 2.3. pKCSM tool interface.
CHAPTER 3. RESULTS AND DISCUSSION
3.1. Molecular docking simulation
3.1.1. Redock 03Q
Before docking and screening the compounds, the co-crystallized ligand 03Q of the 3pp0 crystal was re-docked into the active site to determine the root mean square deviation (RMSD). From there, the suitability of the docking process was assessed. The co-crystallized ligand obtained after re-docking using Autodock Vina was compared with the co-crystallized ligand before re-docking using Chimera software. The RMSD value obtained was 1.076 Å < 1.5 Å, which showed that the docking process and results were suitable and reliable ( Figure 3.1 ).
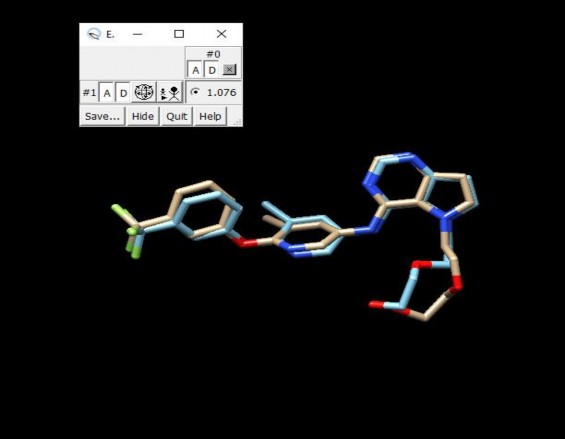
Figure 3.1. RMSD of 03Q co-crystallization before and after redock
The binding energy result when re-docking 03Q ∆G = -10.7 kCal/mol. The interaction between the co-crystallized ligand and 3pp0 is shown in Figure 3.2. The co-crystallized ligand shows the formation of bonds with many important amino acids in the active site such as: -alkyl bonds with LYS753, VAL 734, ALA751,
MET774, LEU 785, LEU796; -σ with LEU852, LEU726; hydrogen bond with GLN799, MET801, ASP863; - with PHE1004; Halogen bond with GLU770; Van der Waals bond with ASN850.
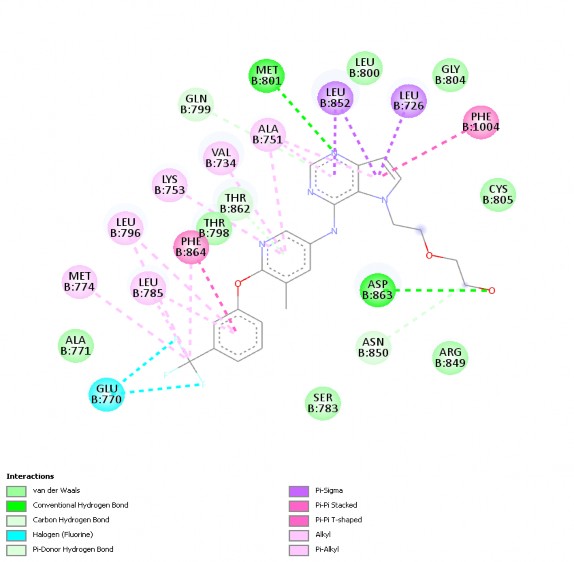
Figure 3.2. 2D interaction of co-crystallized ligand with 3pp0
Figure 3.3. shows the 2D interaction of the co-crystallized ligand with 3pp0 after re-docking. It can be seen that, after re-docking, 03Q still binds to important amino acid groups such as LYS753, VAL734, ALA751, GLN799, MET801, LEU852, LEU726, PHE1004, GLU770, MET774, LEU785, LEU796 or
Van der Waals interaction with ASP863. The interaction of 03Q re-dock differs only from before re-docking in that 03Q re-dock does not show the amino acid ASN850. This result shows the good interaction level of ligand with protein when re-docked.
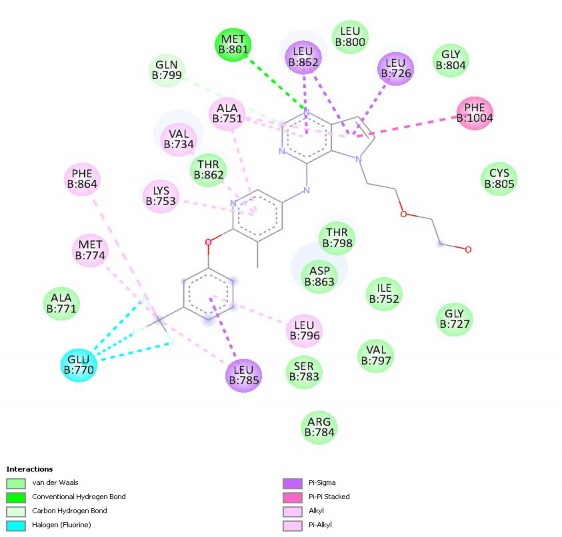
Figure 3.3. 2D interaction of co-crystallized ligand with 3pp0 after re-docking.
Thus, the evaluation of the main RMSD index along with the ability to interact with similar amino acids between the original 03Q structure and the re-docked 03Q confirmed that the docking process was suitable for continuing the docking process of 60 Isoflavone compounds in the database and the positive control compound.
3.1.2. Searching for potential substances from docking results
After the re-docking process proved suitable, docking of 60 Isoflavone compounds and positive controls into the active site of the protein at coordinates (x,y,z) (34, 46, -12) in a 30 Å x 30 Å x 30 Å box [68]. The binding energies ∆G of the 60 compounds and Trastuzumab are shown in Table 3.1 .